
多维度案例 | Mol Cancer:CREPT通过重塑染色质结构驱动三阴性乳腺癌转移
三阴性乳腺癌(triple-negative breast cancer,TNBC)由于缺乏ER、PR和HER2靶点,是目前乳腺癌中治疗选择最有限、预后最差的亚型之一。临床上,TNBC患者往往在早期即出现远处转移,转移而非原发肿瘤本身,是造成患者死亡的主要原因。尽管近年来对TNBC的分子分型、免疫治疗和化疗联合策略不断推进,但一旦发生转移,疗效依然极为有限。
从机制研究角度看,TNBC转移并非由单一突变事件驱动,而是一个涉及基因组变异、表观调控、转录重编程和微环境适应的复杂过程。大量研究已关注 TP53、PI3K-AKT 等经典通路,但这些改变并不能完全解释TNBC中“为何某些肿瘤细胞会获得强烈转移能力“。尤其值得注意的是,体细胞拷贝数变异在TNBC中高度常见,却长期被视为背景噪音,其如何具体塑造转移相关转录程序,仍缺乏系统性解释。
在肿瘤转移这一动态过程中,拷贝数变异是否能够通过重塑染色质高级结构,系统性驱动转移基因网络?又是否存在关键“结构调控因子”承担这一角色?

2025年6月10日,清华大学常智杰、重庆金凤实验室李俊和清华大学王银银共同通讯在《Molecular Cancer》在线发表题为“CREPT is required for the metastasis of triple-negative breast cancer through a co-operational-chromatin loop-based gene regulation”的研究论文。研究通过多组学和三维基因组学手段发现,CREPT在转移性TNBC中发生拷贝数扩增并高表达,是推动TNBC转移的关键因子。机制上,CREPT并非简单作为转录因子,而是通过介导一种由“增强子–启动子环”和“启动子–终止子环”协同构成的染色质高级结构,高效驱动转移相关基因的持续转录。破坏这一结构,无论在细胞水平还是多种小鼠模型中,均可显著抑制TNBC转移,提示CREPT及其介导的染色质结构具有潜在治疗价值。
研究思路与技术路线
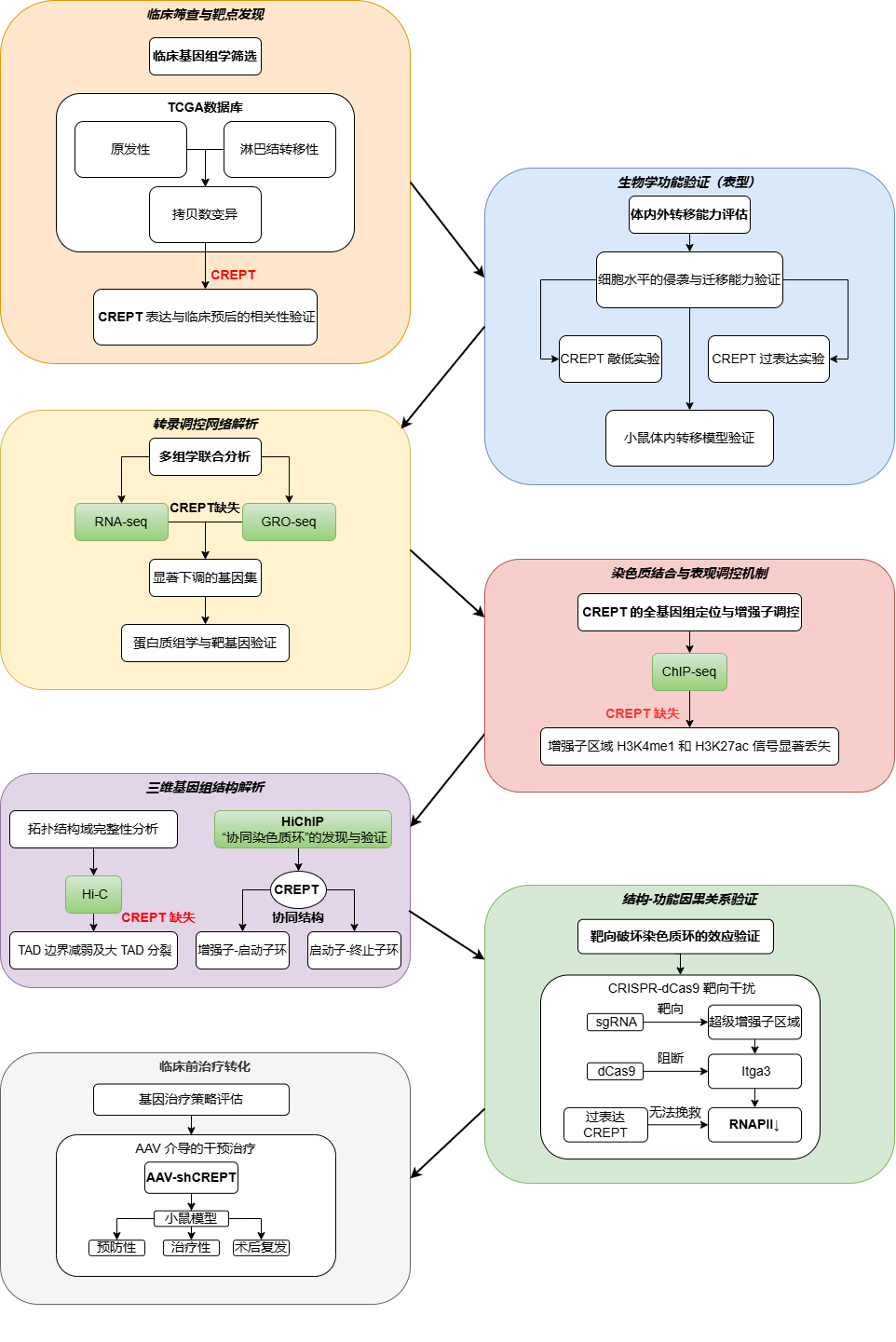
实验设计
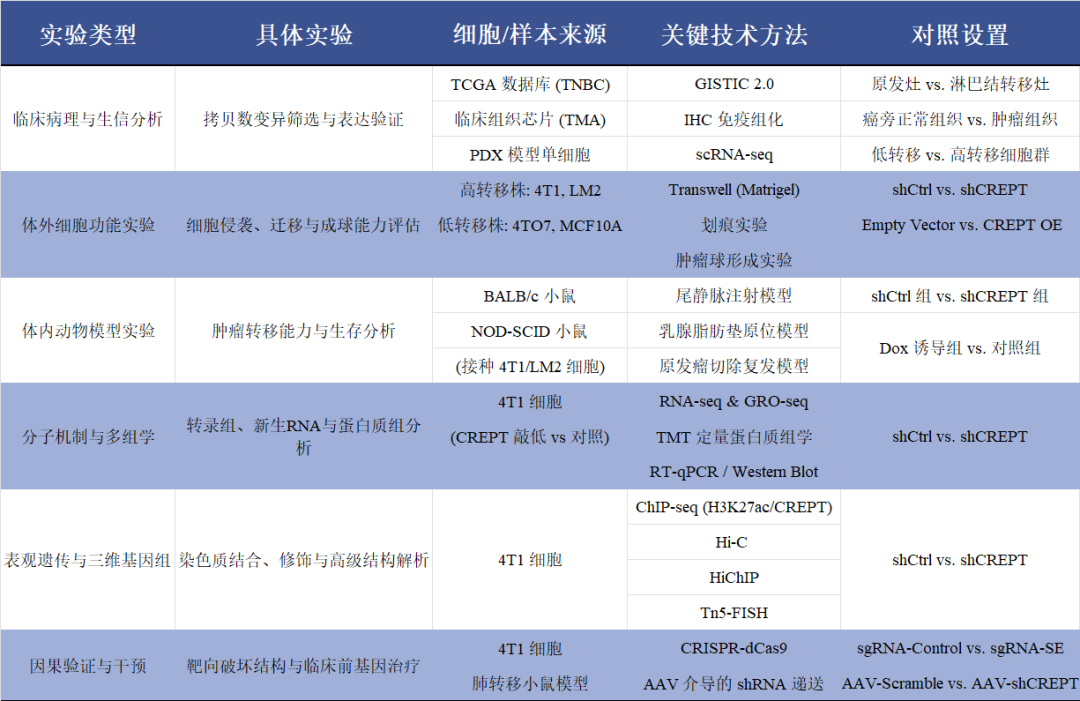
关键研究结果
1、CREPT拷贝数扩增与三阴性乳腺癌转移高度相关
研究首先从临床肿瘤基因组学角度系统筛选TNBC转移驱动因子。通过对TCGA中原发TNBC与淋巴结转移(LNM)TNBC的体细胞拷贝数变异进行比较,研究发现20q11.23是在转移样本中最显著扩增的染色体区域。其中CREPT(RPRD1B)在所有候选基因中表达升高最为显著,其拷贝数扩增与mRNA表达水平高度正相关,并显著预测TNBC患者的不良生存结局。进一步结合单细胞转录组和组织芯片IHC数据,研究明确指出CREPT高表达肿瘤细胞构成TNBC中具有高度转移潜能的亚群,从而在临床与细胞层面确立了CREPT作为转移驱动基因的地位。
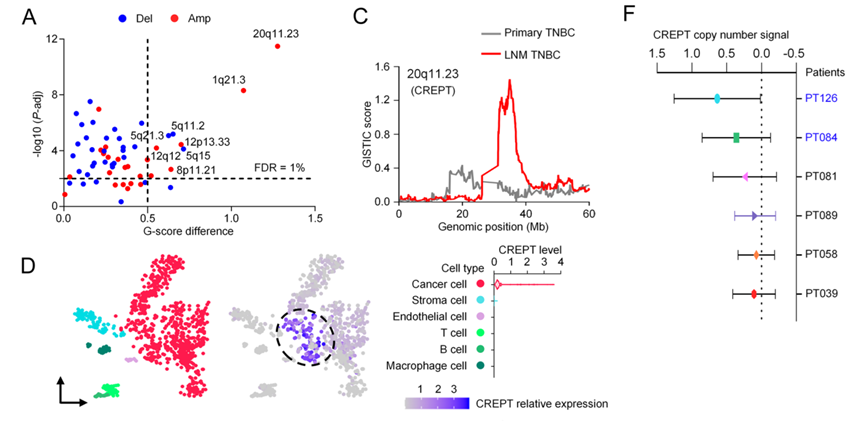
图1. A)基于TCGA数据比较原发TNBC与LNM-TNBC的SCNV图谱,红点代表扩增区域,20q11.23显示最强正向选择信号,提示该区域在转移过程中被选择性扩增;C)GISTIC 分析显示CREPT在LNM-TNBC中的拷贝数显著高于原发肿瘤,直接证明CREPT的基因扩增事件与转移状态密切相关;D–F)单细胞RNA-seq结果显示CREPT高表达细胞集中分布于具有高转移潜能的患者样本中,并伴随明显的染色体拷贝数扩增特征,提示CREPT高表达并非普遍现象,而是转移相关细胞群的特征
2、CREPT在体内外模型中直接驱动TNBC转移
在功能层面,研究通过功能验证实验系统验证 CREPT 对 TNBC 转移的决定性作用。CREPT 在高转移能力 TNBC 细胞系中高表达,敲低 CREPT 显著抑制细胞侵袭、迁移和肿瘤球形成能力,而在低转移细胞中过表达 CREPT 则足以诱导强烈的转移表型。此外,在多种小鼠模型中,无论是尾静脉注射还是原位肿瘤模型,CREPT 的缺失几乎完全阻断远端转移,而不依赖于原发肿瘤生长速度,提示CREPT是TNBC转移必要的驱动因子。
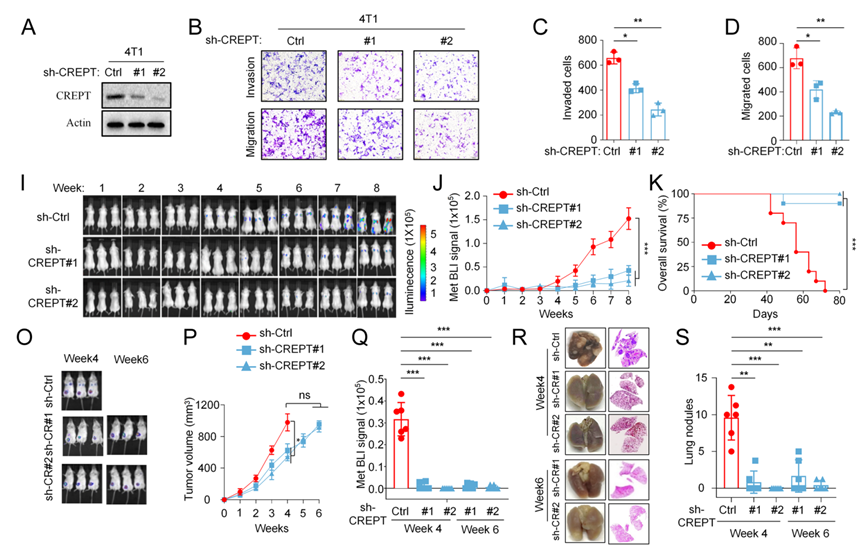
图2. A–D)在4T1高转移细胞中敲低CREPT后,Transwell侵袭和迁移实验显示细胞跨膜能力显著下降,表明CREPT对细胞运动性具有直接调控作用;I–K)尾静脉注射模型结合生物发光成像(BLI)显示,CREPT缺失显著减少多器官转移负荷,并显著延长无转移生存期;O–S)原位乳腺脂肪垫模型中,在原发肿瘤大小相当的条件下,CREPT缺失组几乎不产生肺转移,证明其对转移的调控独立于肿瘤增殖。
3、CREPT广泛上调转移相关基因转录程序
为解析CREPT驱动转移的分子基础,研究结合GRO-seq、RNA-seq与定量蛋白质组学,对CREPT调控的转录网络进行了多层次解析。CREPT缺失导致数千个基因在新生RNA、成熟 mRNA 及蛋白水平同步下调,其中高度富集于细胞黏附、炎症因子、血管生成和ECM重塑等转移核心通路。这一结果表明CREPT并非调控单一转移基因,而是作为上游枢纽,系统性维持转移相关转录程序。
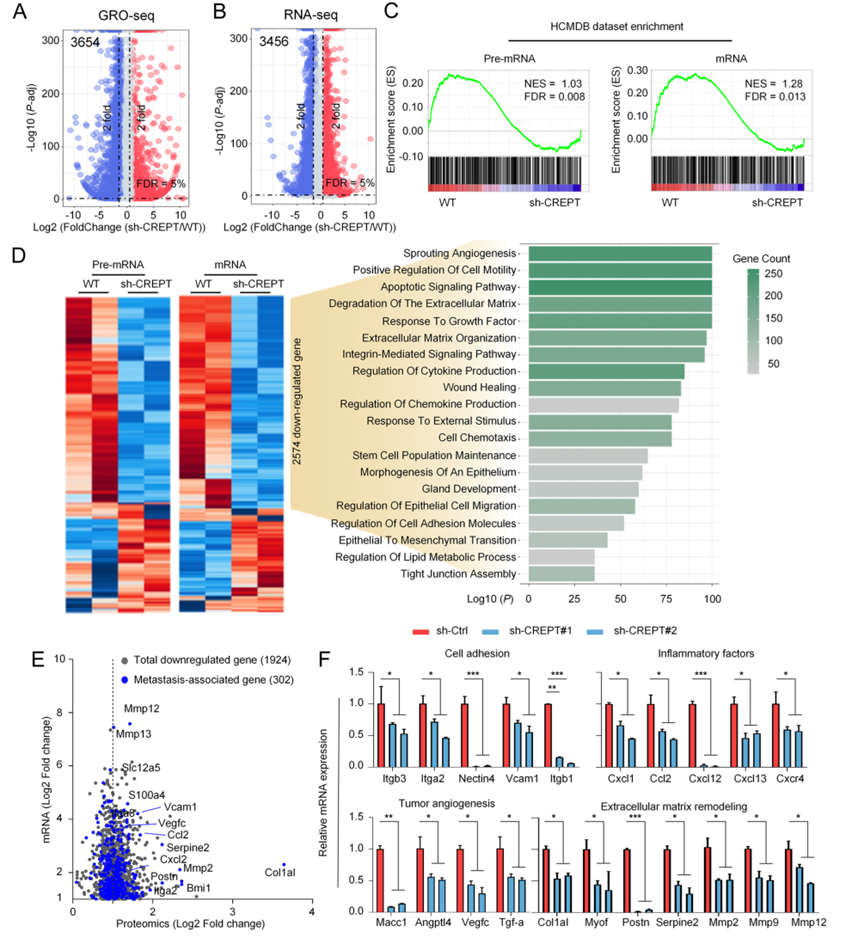
图3.A–B)GRO-seq与RNA-seq火山图显示CREPT缺失后大量基因转录活性显著降低,且两种数据高度一致;D)交叉分析确定2574个同时在nascent RNA与mRNA层面受CREPT调控的基因,并在GO分析中显著富集转移相关生物过程;E)TMT蛋白质组学进一步验证CREPT缺失导致关键转移蛋白(如MMP13、VCAM1、CCL2)显著下调;F)RT-qPCR验证多个转移核心基因在CREPT缺失后显著下调,强化其对转移转录程序的系统性调控作用。
4、CREPT通过p300激活增强子与超级增强子
机制上,研究发现 CREPT 主要占据活跃增强子和超级增强子区域,并通过与 p300 相互作用维持 H3K27ac 水平和染色质开放性。CREPT 缺失显著削弱增强子尤其是超级增强子的结构完整性和活性,提示其通过表观遗传层面放大转移相关基因的转录输出。
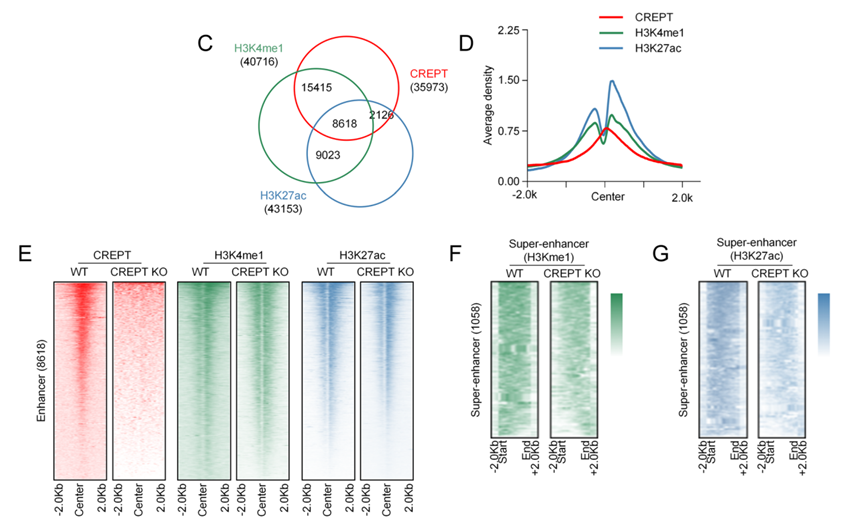
图4.C–E)ChIP-seq显示CREPT与H3K4me1、H3K27ac在增强子区域高度共定位,且CREPT缺失显著削弱这些标志;F–G)在超级增强子区域,CREPT缺失导致H3K4me1与H3K27ac覆盖范围和强度显著下降,表明其对高阶转录调控单元尤为关键。
5、CREPT维持TAD边界稳定性并影响基因表达
在三维基因组层面,Hi-C分析显示CREPT缺失并不显著改变A/B compartment,但会引发大规模TAD分裂和边界减弱。这些结构变化直接影响TAD内顺式调控元件与基因的有效接触,从而导致部分转移基因转录下调。
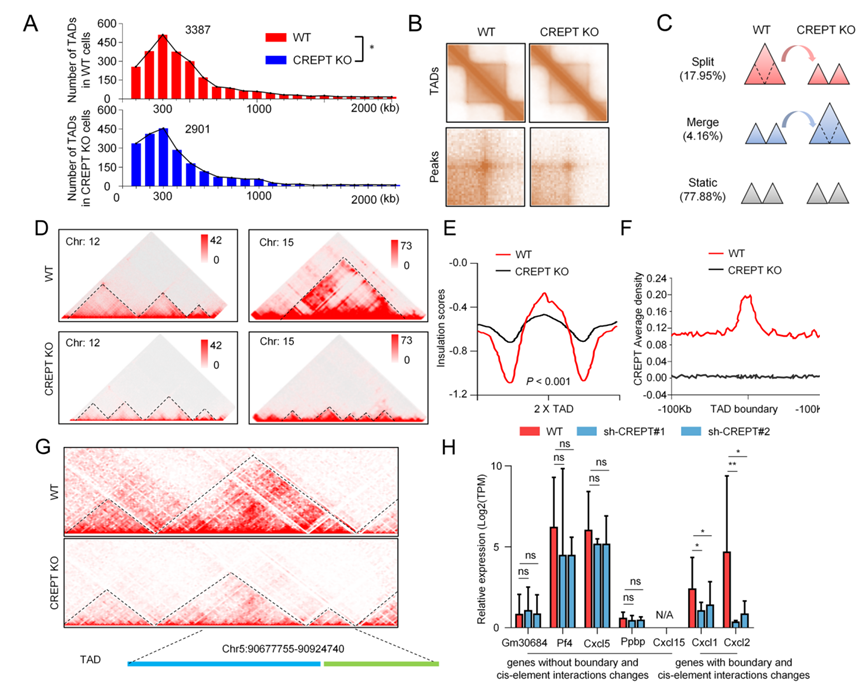
图5.A–C)CREPT缺失后TAD数量减少,且大尺寸TAD显著分裂,提示其对高阶染色质结构稳定性的重要作用;E–F)CREPT 在TAD边界富集,其缺失导致绝缘评分下降,边界功能受损;H)TAD边界受损的基因表达显著下调,而未受影响的基因表达保持稳定,建立结构—功能关联。
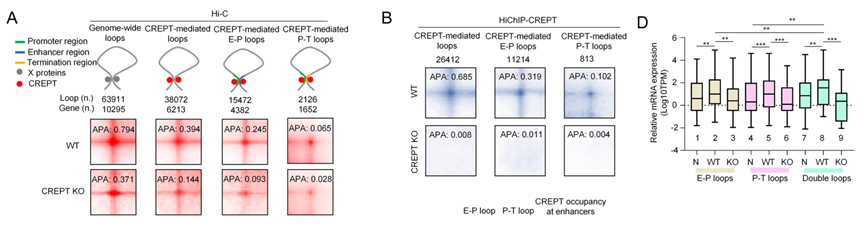
6、CREPT介导增强子–启动子与启动子–终止子染色质环
进一步解析发现CREPT是染色质环形成的核心调控因子,尤其介导增强子–启动子(E–P)环与启动子–终止子(P–T)环。Hi-C与HiChIP数据一致显示,CREPT缺失导致大多数此类环结构强度显著降低,直接影响基因转录效率。
7、CREPT构建“协同染色质环”并高效驱动转移基因表达
研究提出并定义了一种新的高效转录调控结构——协同染色质环(co-operational loops),即E–P环与P–T环在同一基因位点协同存在。全基因组分析鉴定出1082个由协同环调控的基因,其表达水平显著高于仅具有单一环的基因,且高度富集转移功能。CREPT缺失优先破坏此类结构并强烈抑制转移基因表达。
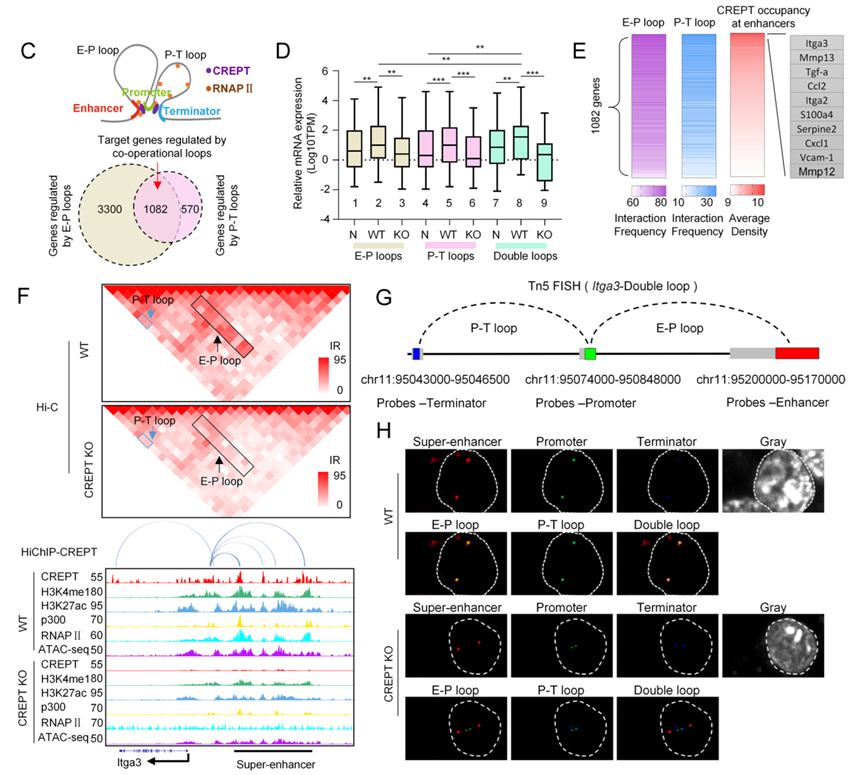
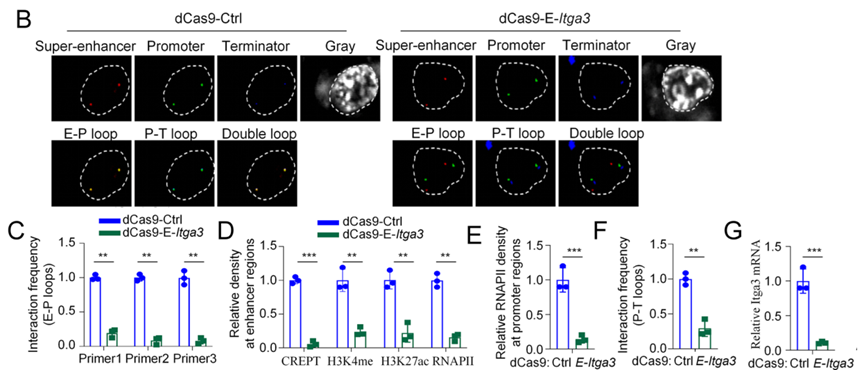
8、协同染色质环协调RNAPII装载与循环并决定转移能力
通过CRISPR-dCas9靶向破坏超级增强子,研究证明协同环是RNAPII高效装载与循环的结构基础。破坏协同环导致RNAPII招募下降、转录中断,并显著抑制体内外转移。即使在CREPT过表达条件下,协同环的破坏仍完全阻断转移基因激活,凸显其功能不可替代性。
研究意义与机制模型
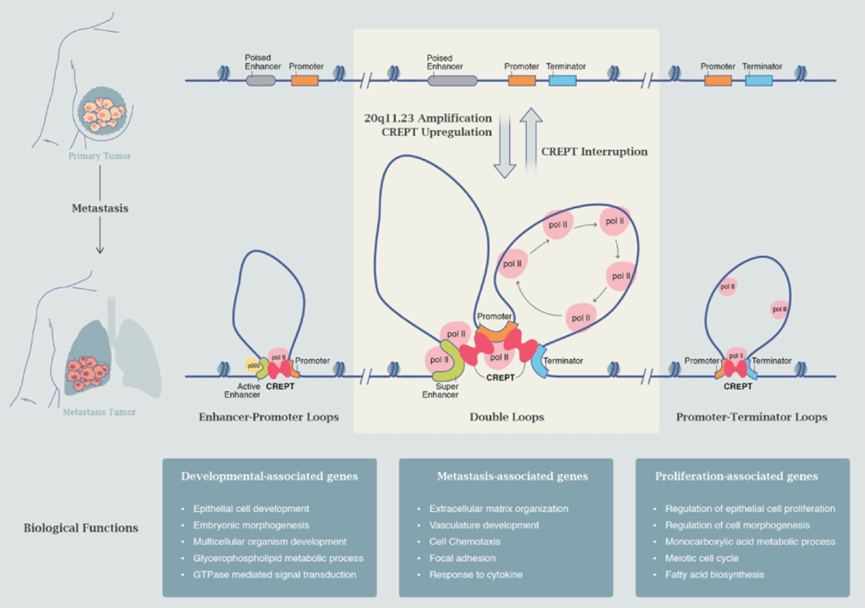
这项工作提出了一个重要概念:体细胞拷贝数扩增并不只是提高基因表达“剂量”,而是可能通过重塑三维基因组结构,系统性重编程转录网络。CREPT的拷贝数扩增,使其成为一种“结构调控枢纽”,而非单一通路分子。研究强调其发现的“协同染色质环”并非对传统增强子–启动子模型的否定,而是在此基础上的拓展:增强子–启动子环负责RNAPII装载,而启动子–终止子环促进RNAPII循环再利用,两者协同,显著提高转录效率。这一模型为理解肿瘤中“持续高表达”的转移基因提供了新的结构性解释。在治疗层面,研究提示:靶向转录结构本身,而非单个转移基因,可能更适合应对TNBC这类高度异质性的肿瘤。无论是通过抑制CREPT,还是未来发展针对协同染色质环的干预策略,都为转移性TNBC的治疗提供了新的思路。


