
表观转录组研究方案
1.1 m6A修饰
m6A是一种动态可逆的修饰方式,在转录后调控中发挥作用,其在调控基因表达、剪接、RNA编辑、RNA稳定性、控制mRNA寿命和降解、影响蛋白翻译[1]等方面扮演重要角色,具有非常重要的研究意义。
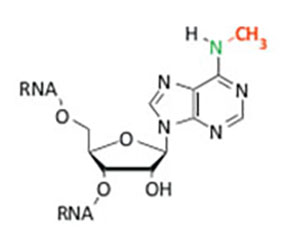
图1. m6A的化学结构示意图
m6A甲基化修饰是由一个多蛋白复合物介导产生的(如图1),目前已知这个复合物的成分包括METTL3、METTL14和WTAP;而负责擦除甲基化修饰基团的是去甲基化酶FTO和ALKBH5。在细胞核中的 HNRNPC负责识别m6A修饰基团,并介导mRNA前体的选择性剪接(可变剪切)。而另外一个m6A识别蛋白HNRNPA2B1[2]则促进pri-miRNA加工成 pre-miRNA。
在细胞质中,不同的m6A位点识别蛋白(reader)介导不同的功能,涉及mRNA翻译、稳定性、剪接以及出核等方面。YTHDF1和YTHDF3[3]识别m6A修饰mRNA, 通过与起始因子及核糖体相互作用促进蛋白质翻译,eIF3识别蛋白也会直接绑定到mRNA 5'UTR 端的m6A位点参与翻译起始。而另外一个识别蛋白YTHDF2与m6A识别将导致mRNA的降解,而YTHDF3与YTHDC2也有类似的功能;而IGF2BP1/2/3则对翻译的稳定性有促进作用;YTHDC1与mRNA剪切以及出核相关。目前还存在其他未知的m6A识别蛋白,相关研究的继续开展将有利于解开m6A在mRNA运输、翻译和存储等功能方面的谜团。

图2. m6A修饰及相关因子[4]
1.2 ac4C修饰
N4-acetylcytidine (ac4C),N4位乙酰胞嘧啶,是真核原核生物中保守的化学修饰,早期研究认为ac4C主要存在tRNA和18S rRNA上[6]。而近期研究显示,mRNA上也存在大量的ac4C,该修饰在促进蛋白翻译,影响RNA稳定性和可变剪接,调控基因表达发挥重要作用,有望成为表观转录组学的新兴发展方向[7,8]。而近期研究显示,mRNA上也存在大量的ac4C,其丰度甚至不低于mRNA携带的m7G帽子修饰[9,10]。NAT10是目前鉴定的唯一同时具有乙酰化酶结构域和RNA结合结构域的蛋白,因此被认为是RNA ac4C修饰酶。
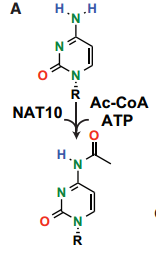
图3. ac4C的化学结构示意图
2018年Arango等人的研究[11]首次揭示mRNA上存在大量ac4C修饰,并且ac4C影响mRNA的稳定性与翻译效率。
目前已鉴定到的核糖核苷修饰已超过140种,大量研究集中在tRNA和rRNA上,而mRNA上的化学修饰及其作用,研究尚处起步阶段。
2.1 研究目标
利用高通量测序技术/三代测序技术,在细胞水平上鉴定RNA修饰及其相关因子对该疾病的影响作用,并从RNA可变剪切、RNA翻译、RNA稳定性三个方面初步探究RNA修饰及其相关因子发挥作用的转录调控机制,再通过组织、细胞、动物水平的分子实验阐明RNA修饰相关蛋白及其介导的RNA修饰失调对疾病发生发展、诊断、治疗及预后的作用。
2.2 研究内容
2.2.1 初步探讨RNA修饰相关蛋白对疾病发生发展的影响
在未确定RNA修饰是否参与疾病发生发展的过程之前,我们首先对是否存在此机制,进行较全面的初筛和初探。
利用与疾病密切相关的细胞株,建立RNA修饰相关因子的敲除/敲降/过表达细胞模型,包括YTHDF1/2/3、FTO、ALKBH5、METTL3/4、YTHDC1、IGF2BP1/2/3。运用SLAM-seq高通量测序技术,对比对照组与敲除/敲降/过表达的转录组稳定性。敲除/敲降/过表达之后RNA稳定性出现显著变化,表明该基因在此过程中发挥着潜在的作用,提示RNA修饰可能通过改变RNA稳定性,影响着疾病的发生发展动态过程,值得深入验证此猜想。
2.2.2 疾病发生发展过程中转录组的RNA修饰鉴定
RNA修饰现象发生于RNA分子上,会发生RNA修饰水平的变化。
a. 运用meRIP-seq高通量测序技术,对正常细胞/组织与病理细胞/组织分别进行m6A甲基化修饰图谱的对比,获得差异m6A修饰位点信息,鉴定m6A修饰在病理过程的存在。
b. 运用acRIP-seq高通量测序技术,对正常细胞/组织与病理细胞/组织分别进行ac4C修饰图谱的对比,获得差异ac4C修饰位点信息,鉴定ac4C修饰在病理过程的存在。
2.2.3 初步解析RNA修饰对疾病发生发展相关基因的转录调控
2.2.3.1 RNA修饰对RNA翻译的影响分析
为了进一步阐明RNA修饰影响该疾病发生发展的机制,
2.2.3.2 RNA修饰对RNA可变剪切的影响分析
为了进一步阐明RNA修饰影响该疾病发生发展的机制,
2.2.4 在组织水平探究RNA修饰对疾病发挥作用的分子机制
2.2.4.1 临床组织样本中的RNA修饰相关蛋白表达情况的分析
利用RNA修饰相关蛋白的抗体,对正常人组织样本与疾病患者组织样本分别进行免疫组化(Immunohistonchemistry)分析,观察该蛋白在两者之间是否存在表达差异,鉴定该蛋白的疾病表达图谱。
2.2.4.1 临床组织样本中的RNA修饰相关蛋白与临床症状关联分析
将免疫组化的蛋白在正常与患者组织临床样本中的差异表达情况,与该病患的临床症状包括(肿瘤)分期、复发、侵袭/转移情况进行关联分析。样本数量大为宜。
2.2.5 在细胞水平探究RNA修饰对疾病发挥作用的分子机制
2.2.5.1 建立RNA修饰相关因子的功能缺失性细胞模型
利用与疾病密切相关的细胞株,将上述实验证明的可能在该疾病中发挥作用的RNA修饰相关因子进行敲降/沉默处理。
2.2.5.2 寻找RNA修饰相关基因的下游致病基因/治疗靶点
(1)沉默RNA修饰相关因子对疾病细胞的RNA修饰影响鉴定
a. 运用meRIP-seq高通量测序技术,对对照组与敲降组细胞分别进行m6A甲基化修饰图谱的对比,获得差异m6A修饰位点信息,预测被敲降的RNA修饰相关因子的下游重要致病基因/治疗靶点。
b. 运用acRIP-seq高通量测序技术,对对照组与敲降组细胞分别进行ac4C乙酰化修饰图谱的对比,获得差异ac4C修饰位点信息,预测被敲降的RNA修饰相关因子的下游重要致病基因/治疗靶点。
(2)下游致病基因/治疗靶点的验证
通过荧光定量RT-qPCR,在RNA水平,检测meRIP-seq/acRIP-seq分析预测的下游致病基因/治疗靶点,在对照组细胞和敲降组细胞上的RNA表达情况,是否因该基因的沉默而出现RNA水平的显著表达差异,验证被敲降的基因与预测靶点的关联性。
通过蛋白免疫印迹Western blot,在蛋白水平,检测meRIP-seq/acRIP-seq分析预测的下游致病基因/治疗靶点,在对照组细胞和敲降组细胞上的蛋白表达情况,是否因该基因的沉默而出现蛋白水平的显著表达差异,验证被敲降的基因与预测靶点的关联性。
2.2.5.3 建立RNA修饰相关因子的功能获得性细胞模型
利用与疾病密切相关的细胞株,将上述实验证明的可能在该疾病中发挥作用的RNA修饰相关因子进行过表达处理。
2.2.5.4 揭示RNA修饰相关因子及其介导的RNA修饰对疾病细胞的影响
(1)在疾病细胞株过表达RNA修饰相关因子之后,通过细胞流式分析仪,对对照组与过表达组细胞,分别进行PI染色,检测细胞周期检测,观察RNA修饰相关因子的过表达对细胞周期的影响。
(2)在疾病细胞株过表达RNA修饰相关因子之后,对对照组与过表达组细胞,分别进行细胞划痕实验(Scratch Assay):在培养了单层贴壁细胞的培养皿上,用枪头在细胞生长的中央区域划线,划痕致伤,继续培养细胞,分别在0h、6h、12h、24h时间点在倒置显微镜下观察,观察RNA修饰相关因子的过表达对细胞的运动特性、迁移能力的影响。
(3)在疾病细胞株过表达RNA修饰相关因子之后,对对照组与过表达组细胞,分别进行细胞侵袭实验(Matrigel Invassion Assay),观察RNA修饰相关因子的过表达细胞穿过重建基质膜或Transwell小室的能力,了解该因子对细胞的体内侵袭转移能力的影响。
(4)还可以根据研究实际需求、疾病细胞株的特性选择进行MTT/CCK-8检测、克隆形成检测、细胞凋亡检测、细胞趋化实验等。
3.1 本项目的创新之处
(1)本项目从表观遗传学的角度,研究疾病发生发展过程中表观转录组学的变化,探究RNA修饰在此动态过程发挥的作用;
(2)探究RNA修饰调控转录调控的作用机理,从RNA稳定性、RNA翻译、RNA可变剪切的角度剖析RNA修饰对疾病病理的作用靶点。
(3)深入RNA修饰调控转录调控的作用机理,通过细胞功能获得性实验及功能缺失性实验,找到RNA修饰相关因子的下游致病基因/治疗靶点,揭示RNA修饰相关因子及其介导的RNA修饰对疾病细胞的影响,能为疾病发生发展、诊断、治疗、预后手段的提升提供新的思路。
3.2 拟解决的科学问题
从表观遗传学层面阐明该疾病的病理机制,挖掘疾病发生发展过程中关键的转录调控靶点基因,有望促进治疗药物、治疗手段的开发,或提高疾病早期诊断、预后评估手段的准确性。
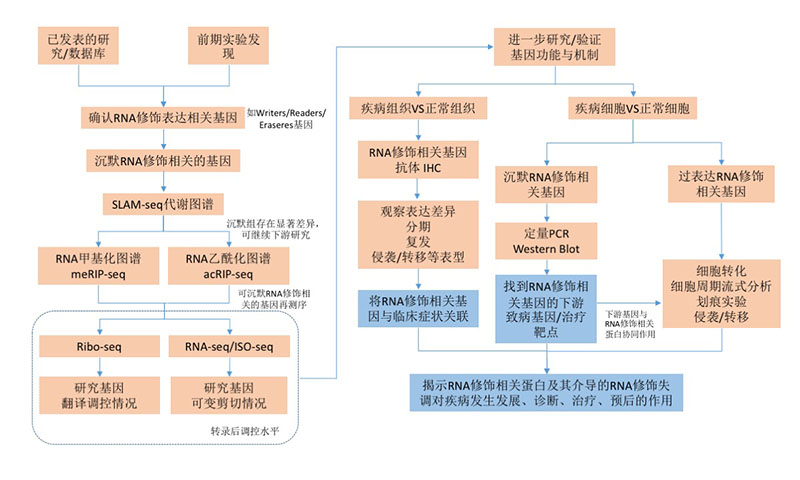
图4. 表观转录组研究技术路径图
5.1 SLAM-seq
技术原理:向合成中的RNA链中掺入4-thiouridine (S4U),使原本的碱基T被S4U修饰,从而在反转录中被错认为C,导致原本应该为碱基A的位置错配成为G。通过测序分析这些错配的G,就可以对新合成mRNA或其他longRNA进行直接的定量。
实验分组及样本要求:利用与疾病密切相关的细胞株,建立RNA修饰相关因子的敲除/敲降/过表达细胞模型,包括YTHDF1/2/3、FTO、ALKBH5、METTL3/4、YTHDC1、IGF2BP1/2/3,此为敲除/敲降/过表达组;与野生型细胞进行比较。1*106个细胞/每样本,在培养的活细胞中掺入S4U碱基。
分析内容:
1. 序列比对
2. 文库质控
3. Mapped Reads过滤
4. 背景T>C SNP校正
5. mRNA或longRNA*异表达分析
6. 靶基因GO富集分析
7. 靶基因KEGG富集分析
8. mRNA或longRNA转录本水平T>C counts计数与表达量化
9. mRNA或longRNA转录本水平Total reads计数与表达量化
10. mRNA或longRNA稳定性分析
11. mRNA或longRNA半衰期计算
* longRNA包括mRNA、lncRNA。
5.2 meRIP-seq
技术原理:基于抗体特异性结合甲基化修饰的碱基的原理,以RNA免疫共沉淀富集甲基化修饰片段为基础,然后通过高通量测序,在全转录组范围内研究发生甲基化的RNA区域,高效获得结果。
实验分组及样本要求:
a. 利用与疾病密切相关的细胞株,建立RNA修饰相关因子的敲除/敲降/过表达细胞模型,包括YTHDF1/2/3、FTO、ALKBH5、METTL3/4、YTHDC1、IGF2BP1/2/3,此为敲除/敲降/过表达组;与野生型细胞进行比较,每组3个或以上样本,5*106~107个细胞/每样本。
b. 正常人细胞/组织与疾病患者细胞/组织作为两组进行对比,每组3个或以上样本,5*106~107个细胞/每样本,或10~20mg组织/每样本。
分析内容:
1、 原始数据过滤及质控
2、参考基因组比对
3、Reads在染色体上的分布
4、Peak Calling 分析
5、Peak 可视化
6、Peak 统计分析
7、m6A 的基本特征
7.1 peak 在基因元件的分布
7.2 reads 在基因元件的分布
7.3 Peak 关联基因的特征
8、差异peak 分析
9、差异peak关联基因GO,KEGG分析
10、 motif分析
高级分析内容:
1. RNA-seq表达差异与m6A修饰差异关联分析
2. RNA可变剪切与m6A修饰差异关联分析
3. 翻译组RIBO-seq翻译差异与m6A修饰差异关联分析
5.3 acRIP-seq
技术原理:基于抗体特异性结合乙酰化修饰的碱基的原理,以RNA免疫共沉淀富集乙酰化修饰片段为基础,然后通过高通量测序,在全转录组范围内研究发生乙酰化的RNA区域,高效获得结果。
实验分组及样本要求:
分析内容:
1、Peak Calling 分析
2、Peak 可视化
3、Peak 统计分析
4、ac4C 的基本特征
4.1 peak 在基因元件的分布
4.2 reads 在基因元件的分布
4.3 Peak 关联基因的特征
5、差异peak 分析
6、差异peak关联基因GO,KEGG分析
7、 motif分析
高级分析内容
8、RNA-seq表达差异与ac4C修饰差异关联分析
9、mRNA可变剪切与ac4C修饰差异关联分析
10、翻译组RIBO-seq翻译差异与ac4C修饰差异关联分析
5.4 Ribo-seq
技术原理:利用低浓度RNase处理核糖体—新生肽链复合物,降解没有核糖体覆盖的mRNA片段,随后再去除核糖体,通过二代测序技术检测被核糖体保护的约22~30 bp的RNA小片段 (称为ribosome footprints,RFPs;又称ribosome protected fragments,RPFs。两者等价),可得到核糖体分布的位置信息,据此可得到每种转录本上核糖体的分布、密度,可推测起始密码子位置(包括非ATG起始)、ORF(开放阅读框)位置、翻译暂停区域以及uORFs (上游开放阅读框)等信息[3]。
实验分组及样本要求:
利用与疾病密切相关的细胞株,建立RNA修饰相关因子的敲除/敲降/过表达细胞模型,包括YTHDF1/2/3、FTO、ALKBH5、METTL3/4、YTHDC1、IGF2BP1/2/3,此为敲除/敲降/过表达组;与野生型细胞进行比较,每组3个或以上样本,1*107个细胞/每样本。
正常人细胞/组织与疾病患者细胞/组织作为两组进行对比,每组3个或以上样本,1*107个细胞/每样本,或300mg组织/每样本。
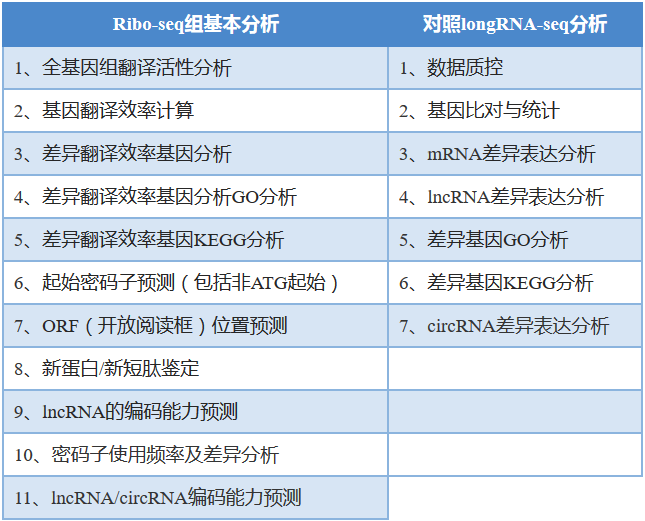
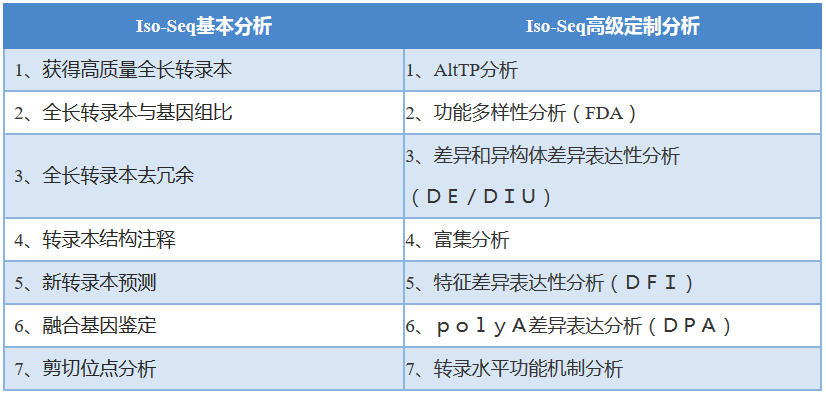
5.5 Iso-Seq
技术原理:PacBio公司推出三代测序仪PacBio RSII,该平台采用单分子实时测序技术,以单分子实时测序反应(single-molecule real-time,SMRT)芯片为载体进行测序,三代测序数据具有读长(read)长(平均8-15kb左右)和无GC偏好性等特点,这些数据特征可以有力弥补了一代和二代测序技术的不足,从而使其具有广泛应用市场。而PacBio的异构体测序(ISO-Seq)采用长读取序列来测序长达10 kb的转录本异构体,是近年来三代测序在RNA领域的热点。其平均读长8-15Kb,可以轻松跨越从 5´末端到 3´-Poly A tail 的完整转录本,从而准确鉴定异构体,可实现对可变剪切(Alternative Splicing, AS)、可选择性多聚腺苷酸化(alternative polyadenylation,APA)、可变转录起始位点(Alternative Transcription Start Sites ,ATSS)、融合基因鉴定、ORF/NMD预测、CDS预测、新转录本发现、功能注释,差异表达和功能多样性分析等内容。
传统二代测序技术分析上,对基因表达数据的功能分析是基于比对的基因而非以转录本为中心,因此短读RNA-seq的解析存在严重局限性,不足以揭示基因表达功能与转录后调控机制,而ISO-Seq测序提供了全新的方法方式去探索RNA世界。
技术优势:
三代测序无需组装可获得准确度大于99%的高质量转录本(HQ high-quality isoforms)
使用最新Iso-Seq3分析流程,获取最新分析手段
使用最新的全长转录本的功能分析(FIT Functional Iso-Transcriptomics analysis)
数据分析多种选择,定制分析内容
实验分组及样本要求:
利用与疾病密切相关的细胞株,建立RNA修饰相关因子的敲除/敲降/过表达细胞模型,包括YTHDF1/2/3、FTO、ALKBH5、METTL3/4、YTHDC1、IGF2BP1/2/3,此为敲降组(沉默组);与野生型细胞进行比较,每组3个或以上样本,5*106~1*107个细胞/每样本,或20ug Total RNA。
正常人细胞/组织与疾病患者细胞/组织作为两组进行对比,每组3个或以上样本5*106~1*107个细胞/每样本,或10mg组织/每样本,或20ug Total RNA。
分析内容:
[1] Zhou C, Molinie B, Daneshvar K, et al. Identification and characterization of m6A circular RNA epitranscriptomes[J]. bioRxiv, 2017: 115899.
[2] Alarcon C R, Goodarzi H, Lee H, et al. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events[J]. Cell, 2015, 162(6):1299~1308.
[3] Yang Y, Fan X, Mao M, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine[J]. Cell Res, 2017, 27(5):626~641.
[4] Cao G, Li H B, Yin Z, et al. Recent advances in dynamic m6A RNA modification[J]. Open Biol, 2016, 6(4):160003.
[5] Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq[J]. Nature, 2012, 485(7397):201~206.
[6] Dong, C., Niu, L., Song, W., Xiong, X., Zhang, X., Zhang, Z., Yang, Y., Yi, F.,Zhan, J., Zhang, H., et al. (2016). tRNA modification profiles of the fast-proliferating cancer cells. Biochem. Biophys. Res. Commun. 476, 340–345.
[7] Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B.M., Strein,C., Davey, N.E., Humphreys, D.T., Preiss, T., Steinmetz, L.M., et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins.Cell 149, 1393–1406.
[8] Arango D, Sturgill D, Alhusaini N, et al. Acetylation of cytidine in mRNA promotes translation efficiency[J]. Cell, 2018, 175(7): 1872-1886. e24.
[9] Dong, C., Niu, L., Song, W., Xiong, X.,Zhang, X., Zhang, Z., Yang, Y., Yi, F.,Zhan, J., Zhang, H., et al. (2016). tRNAmodification profiles of the fast-proliferating cancer cells. Biochem. Biophys.Res. Commun. 476, 340–345.
[10] Castello, A., Fischer, B., Eichelbaum,K., Horos, R., Beckmann, B.M., Strein,C., Davey, N.E., Humphreys, D.T., Preiss,T., Steinmetz, L.M., et al. (2012). Insights into RNA biology from an atlas ofmammalian mRNA-binding proteins.Cell 149, 1393–1406.
[11] Arango D, Sturgill D, Alhusaini N, et al. Acetylation of cytidine in mRNA promotes translation efficiency[J]. Cell, 2018, 175(7): 1872-1886. e24.