
rRPO-seq
rPRO-seq
Rapid Precision nuclear run-on sequencing
Rapid Precision nuclear run-on sequencing (rPRO-seq) represents an optimized PRO-seq workflow. This approach maintains single-nucleotide resolution and enhances both speed and practicality via streamlined adapter design and process optimization. Specifically, it curtails the library preparation time to approximately 15 hours, decreases the minimum input requirement, mitigates adapter-dimer formation, and elevates library efficiency and data quality. Consequently, it is well-suited for clinical materials and rare cell types.
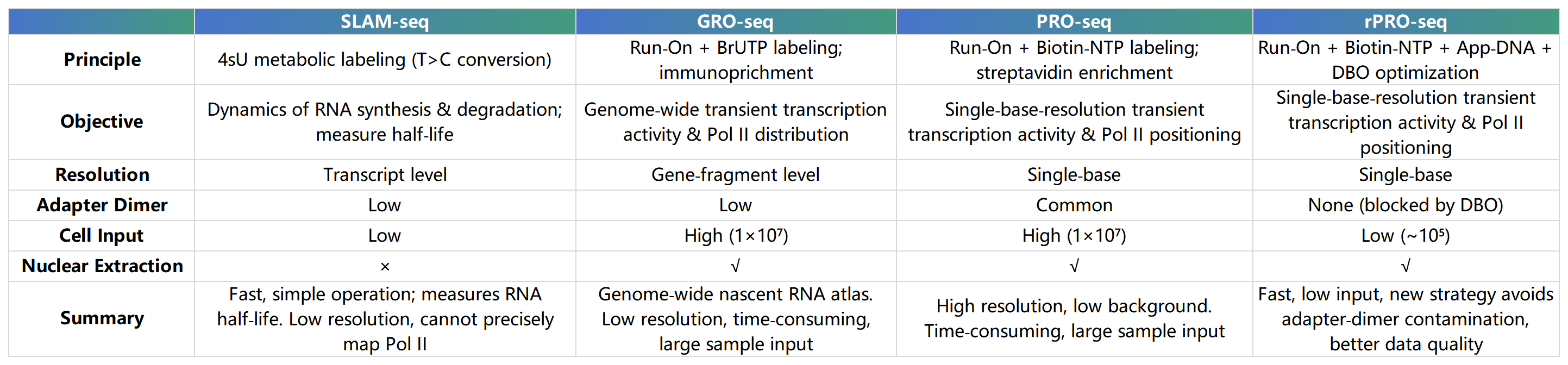
Methods Comparison
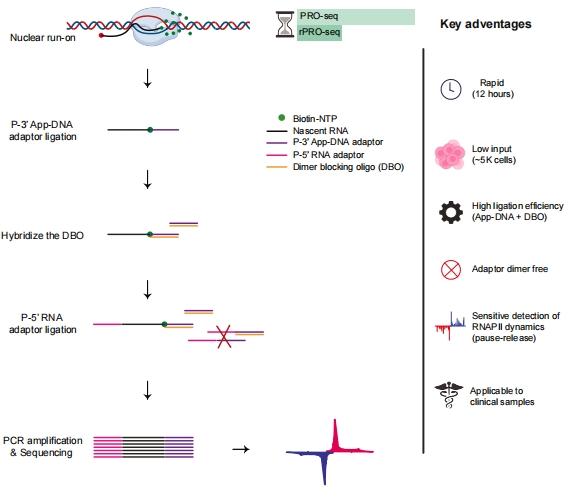
Why rPRO-seq?
Relative to conventional PRO-seq, rPRO-seq provides:
1. Faster turnaround:
library preparation reduced from 4–5 days to ~15 h, accelerating projects and reducing degradation risk.
2. Lower input:
as few as ~10^5 cells (vs 1–2×10^7), broadening sample compatibility.
3. Higher usable yield:
The App-DNA + DBO strategy increases ligation efficiency and minimizes adapter dimers, improving effective data output.
Applications
1. Transcriptional regulation
a. Promoter-proximal pausing: single-nucleotide mapping of RNA Pol II pause sites to study pause-release regulation.
b. Enhancer function: sensitive eRNA detection to support enhancer-promoter network inference.
2. Epigenetic regulation
a. Chromatin–transcription coupling: integrate with ChIP-seq, CUT&Tag, and ATAC-seq to relate TF binding, histone marks, and accessibility to transcriptional activity.
b. Nascent vs steady-state RNA: compare production (nascent RNA) with abundance (steady-state RNA) to estimate RNA turnover or synthesis efficiency and infer post-transcriptional effects.
c. Non-coding transcription: capture unstable species, including upstream antisense transcripts, eRNAs, and lncRNAs.
3. Disease mechanisms and drug development
a. Transcriptional dependency: rapid identification of direct transcriptional targets of epigenetic agents (e.g., BET inhibitors).
b. Response kinetics: distinguish primary transcriptional effects from secondary adaptive programs.
c. Neurodevelopmental studies: resolve transcriptional programs regulated by key neuronal factors.
4. Development and stem cell biology
Profile transient transcriptional reprogramming during differentiation and self-renewal in embryonic stem cells, tissue stem cells, and rare progenitors (e.g., hematopoietic progenitors).
Sample requirements
Human, mouse, and rat; other species require evaluation
≥ 1×10^5
Cells
> 85%
Viability
Data Analysis
Basic analysis:
1. Adapter trimming and low-quality read filtering
2. Genome alignment
3. Transcriptionally active region calling
4. Activity classification for promoters and gene bodies
5. Regional read counting and summary statistics
6. TSS-centered metagene profiling
7. Traveling Ratio: promoter-to-gene-body signal ratio (elongation efficiency)
8. Traveling Matrix: 2D classification of transcriptional-state shifts (four-quadrant analysis)
9. Differential analysis
10. GO/KEGG enrichment analysis
Advanced analysis:
1. eRNA analysis
2. Customized: fee-based, subject to evaluation
Demo
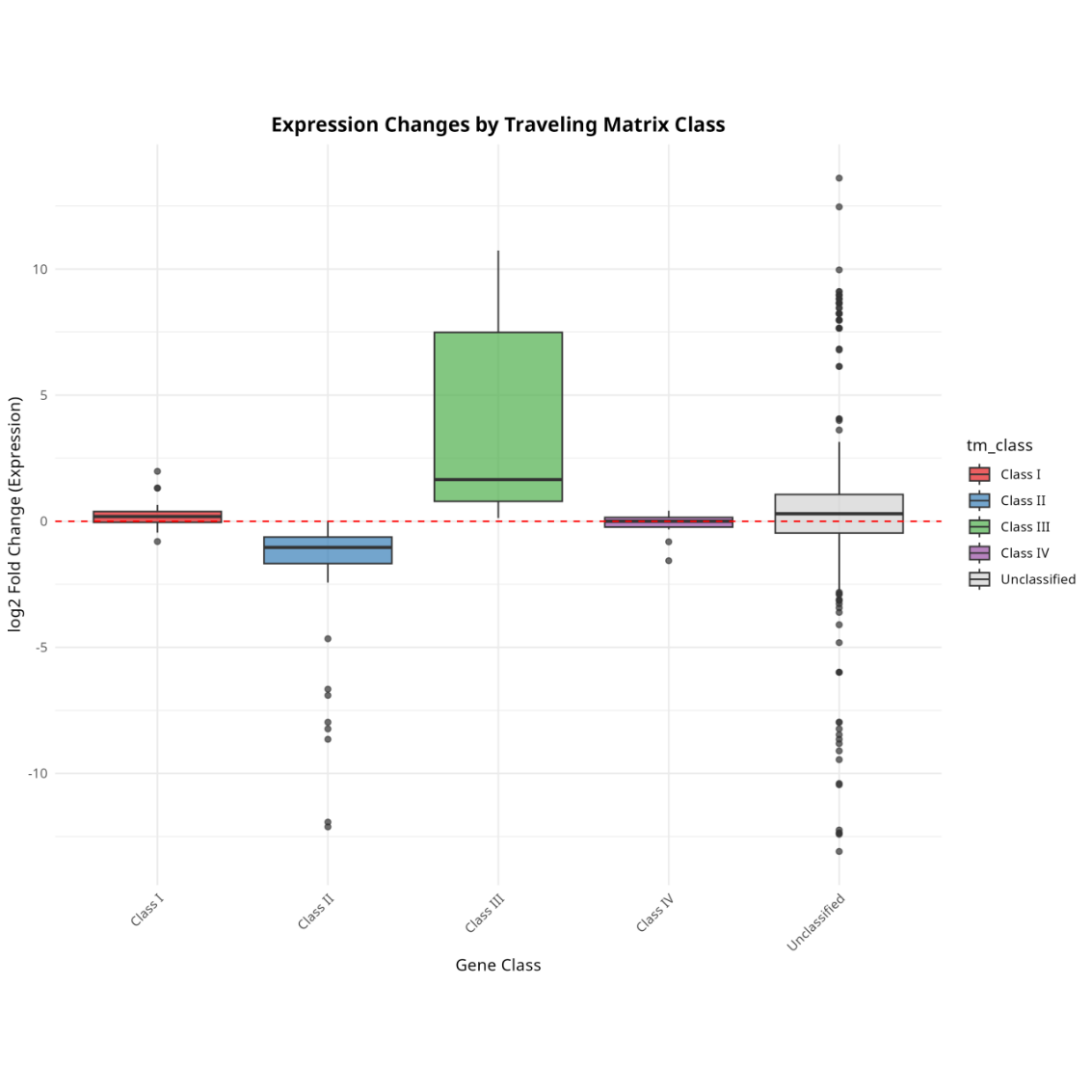
Fold-change distribution across TM classes
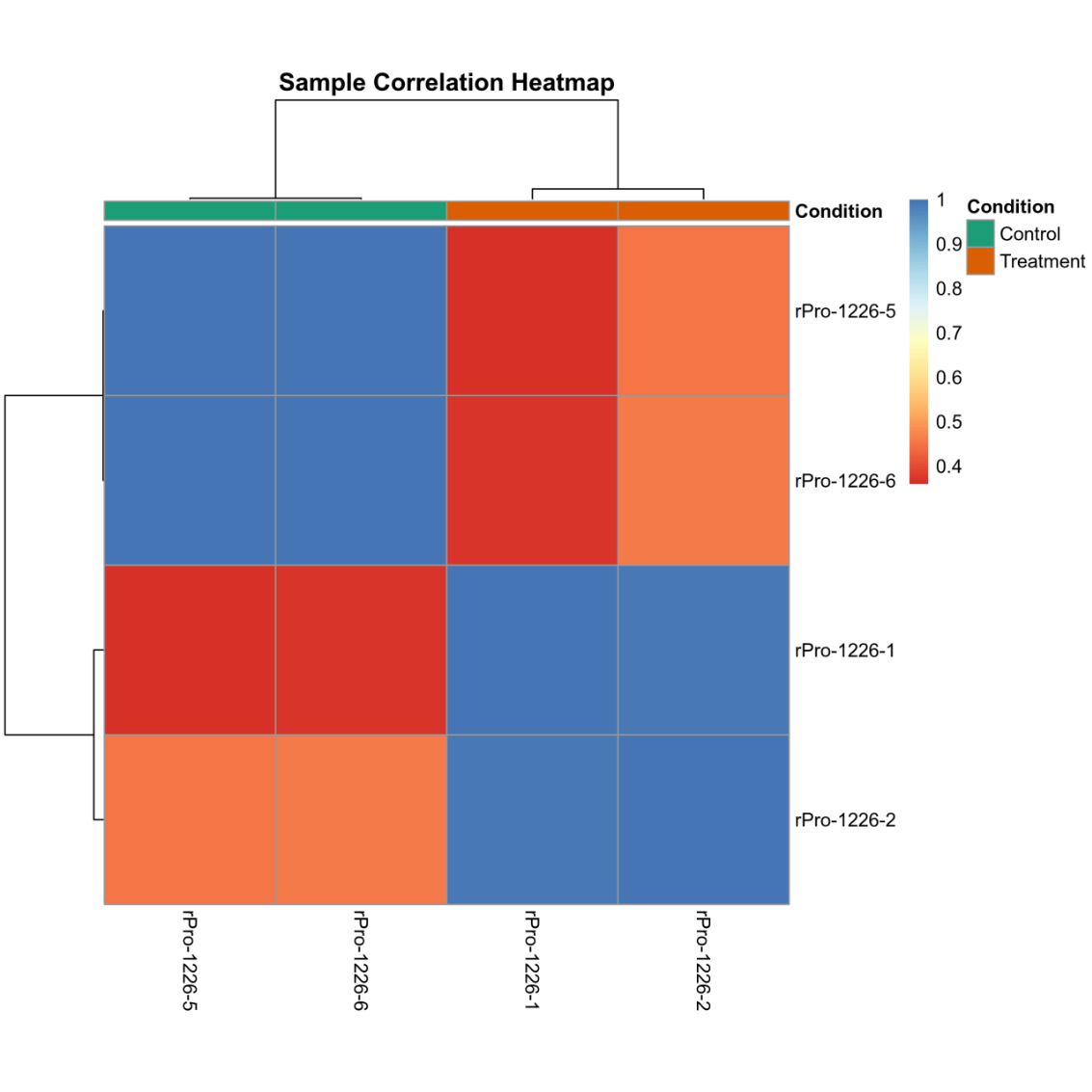
Sample correlation plot
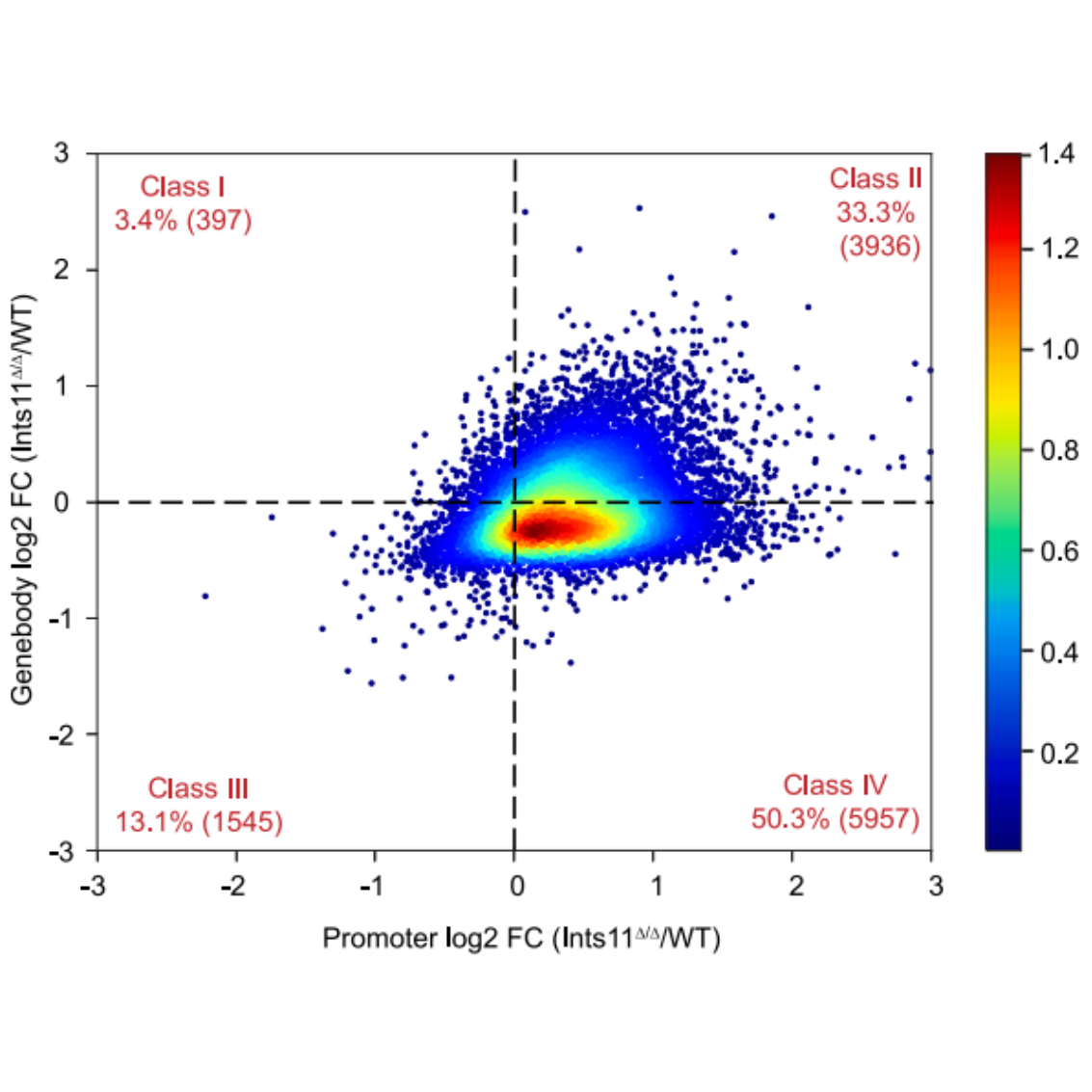
Traveling Matrix classification plot (Cingaram PKR, et al. Mol Cell. 2025.)
Class I: pausing (promoter ↑, gene body ↓);Class II: repression (promoter ↓, gene body ↓);Class III: activation (promoter ↑, gene body ↑);Class IV: pause release (promoter ↓, gene body ↑)
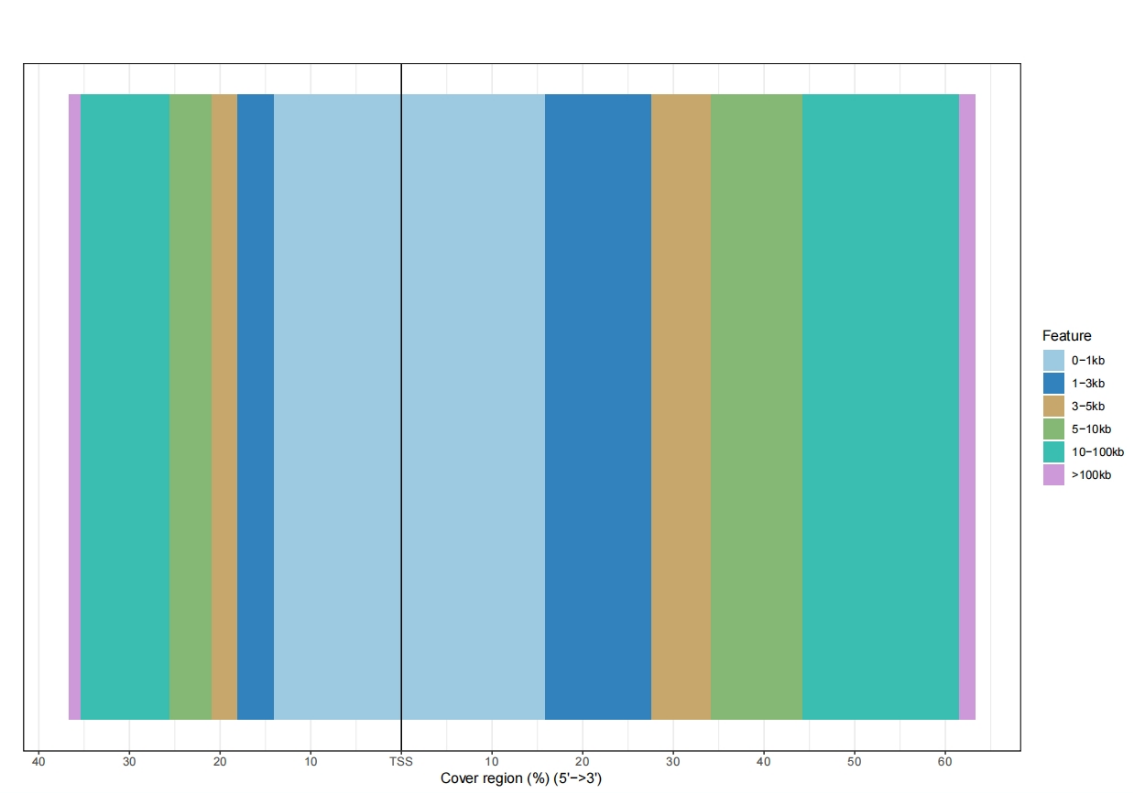
TSS distance distribution
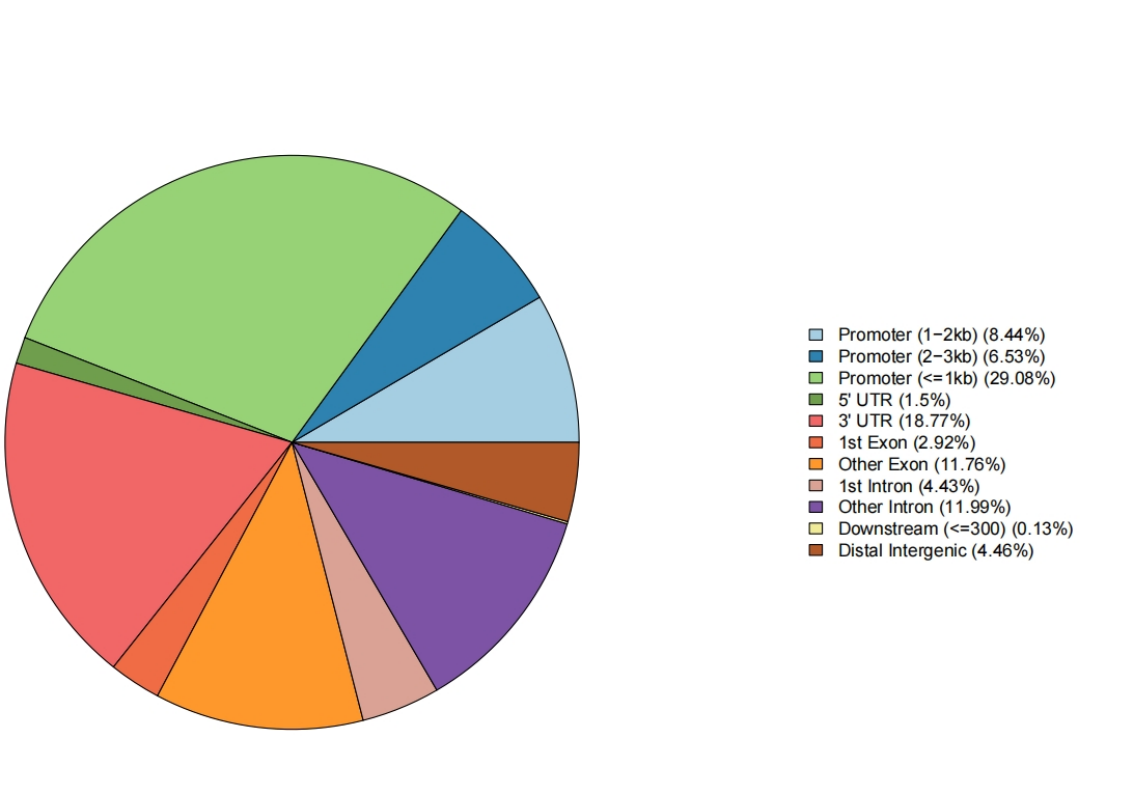
Genomic annotation pie chart
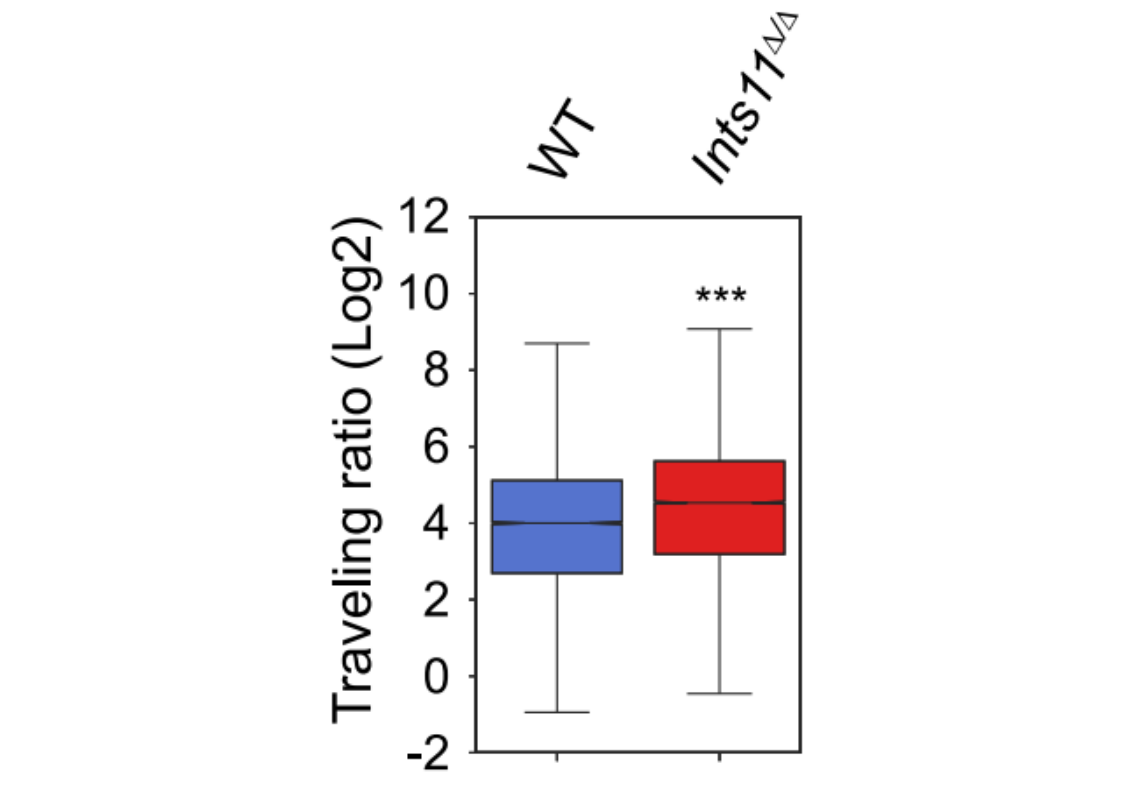
Traveling Ratio analysis (Cingaram PKR, et al. Mol Cell. 2025.)