生物分子凝聚体如何引爆下一代创新药研发?药物开发新纪元的革命性力量与研发路径
生物分子凝聚体(Biomolecular Condensates)作为细胞内一种不依赖膜结构的区室化机制,正迅速成为生命科学和药物开发领域的前沿。这些通过液-液相分离(Liquid-Liquid Phase Separation, LLPS)形成的动态区室,在调控关键生理过程中扮演着核心角色,其功能失调与癌症、神经退行性疾病、心脏病及代谢性疾病等多种重大疾病的发生发展密切相关。下面小编将深入探讨了生物分子凝聚体在药物开发中的巨大应用前景,系统阐述了其作为新型药物靶点的理论基础、关键的疾病关联、创新的药物开发策略,并结合以Dewpoint Therapeutics为代表的行业先驱的最新临床进展,分析该领域面临的挑战与未来发展方向。靶向凝聚体为攻克传统“不可成药”靶点提供了革命性的思路,预示着一个全新的药物发现范式正在到来。
via:youtube
1. 超越传统靶点,聚焦细胞“微反应器”
传统的药物开发遵循“一把钥匙开一把锁”的模式,主要针对具有明确三维结构和结合口袋的蛋白质。然而,人类蛋白质组中大量存在着功能重要但缺乏稳定结构的内在无序蛋白(Intrinsically Disordered Proteins, IDPs),这些蛋白长期以来被视为“不可成药”的靶点【1】。
近年来,对生物分子凝聚体的研究揭示了细胞组织和调控自身生化反应的一种全新方式。凝聚体如同细胞内的“微反应器”,通过相分离过程将特定的蛋白质和核酸等分子富集起来,形成高度浓缩且动态变化的无膜细胞器,从而在时空上精确调控基因转录、信号转导、DNA修复和RNA代谢等核心生命活动【2, 3】。
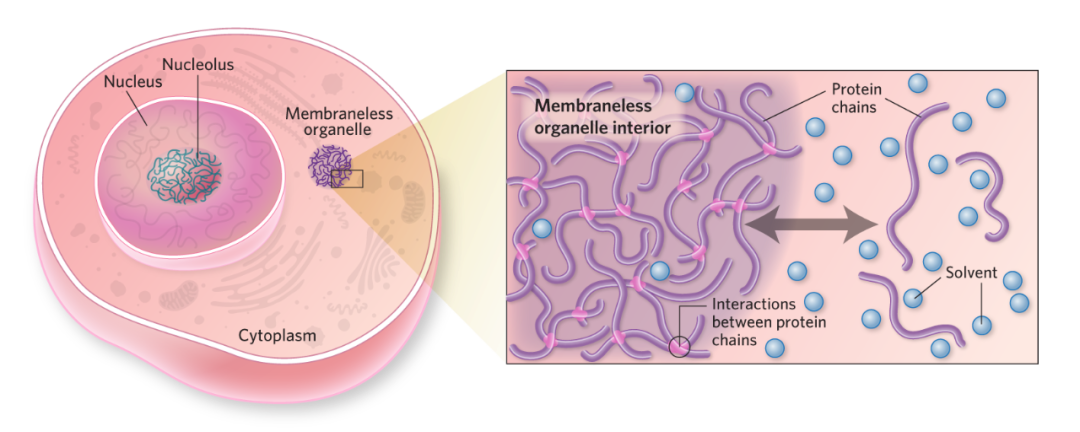
这些凝聚体的形成和解散受到细胞内外信号的精密调控,而其功能的异常,如错误的形成、固化(老化)为不可逆的聚集体、或组分异常,已被证实是多种疾病的根本驱动因素【4】。这一发现为药物开发提供了革命性的新视角:不再仅仅靶向单个蛋白质的活性位点,而是通过调节整个凝聚体的状态(如形成、溶解、物理特性或组分)来干预疾病进程。这种策略为攻克像MYC、TDP-43 等长期困扰药物开发者的“不可成药”靶点开辟了全新的道路。
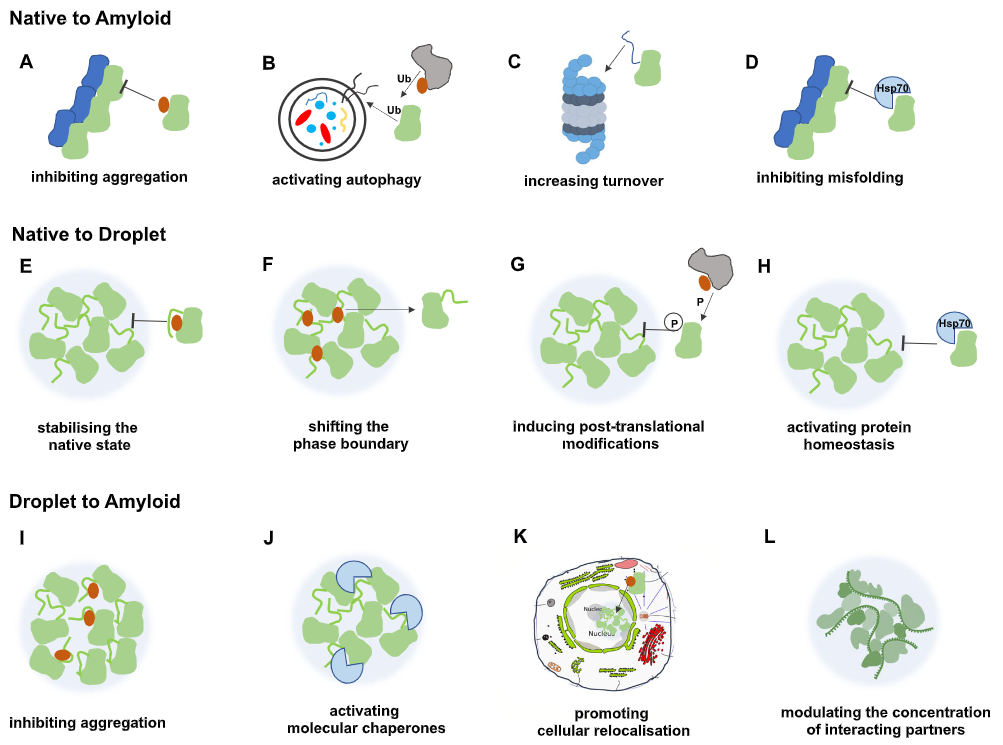
2. 凝聚体功能失调:多种重大疾病的共同根源
凝聚体功能异常与多种人类重大疾病的病理机制紧密相连,使其成为极具吸引力的药物靶点。
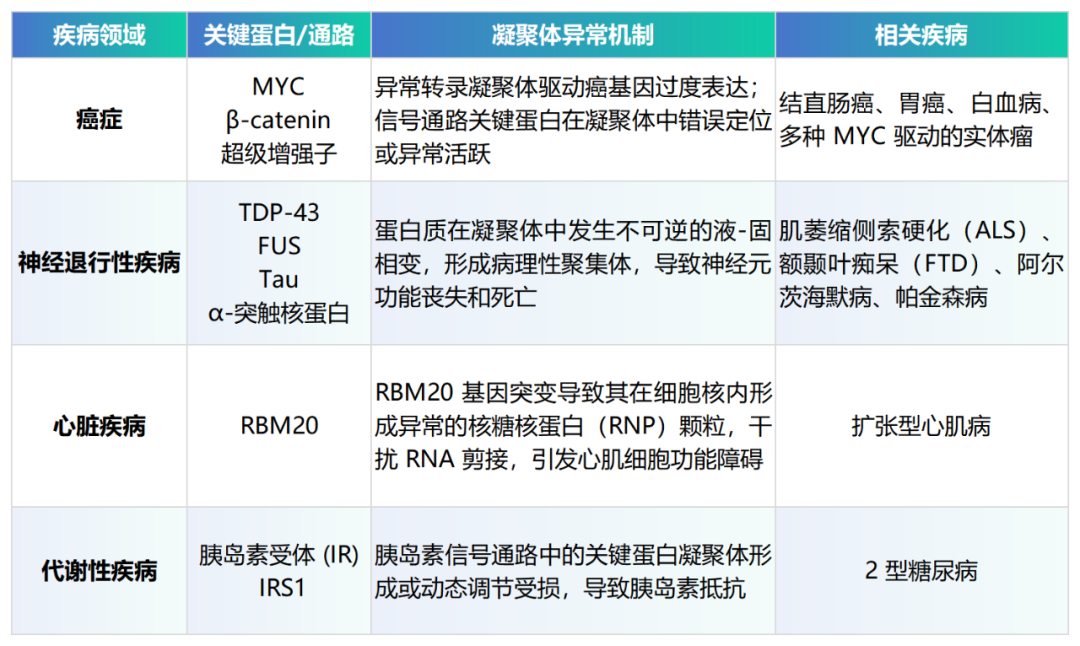
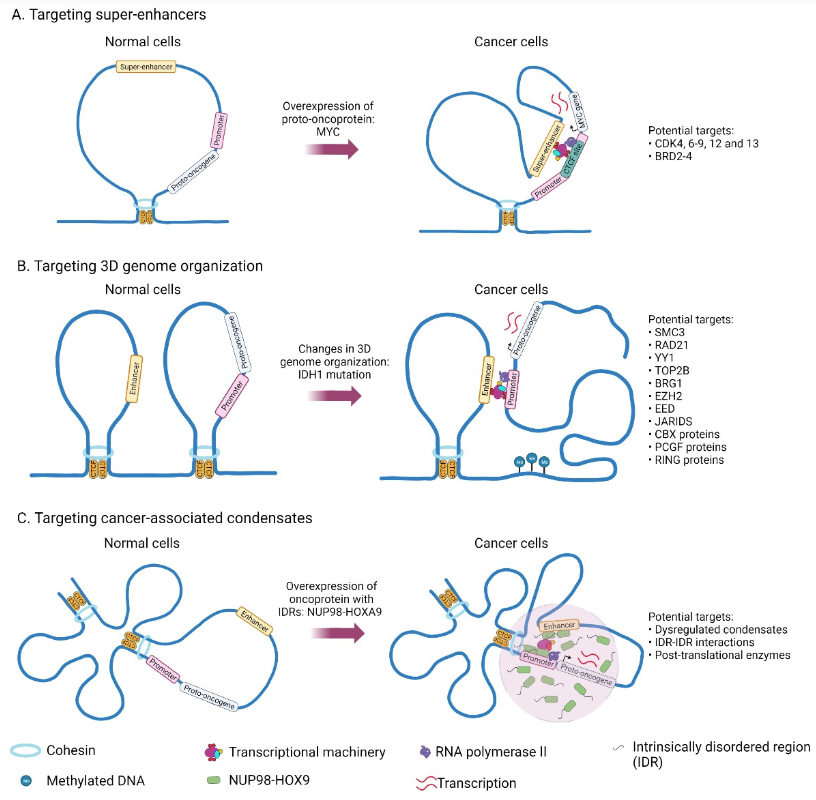
在癌症领域,研究发现许多关键的癌基因(如MYC)和信号通路(如Wnt/β-catenin)的活性高度依赖于转录凝聚体的形成。这些凝聚体在超级增强子区域富集转录因子和RNA聚合酶II,形成一个高效的转录中心,驱动癌细胞的失控性增殖【5】。因此,通过破坏这些致癌凝聚体,有望从根源上关闭癌症引擎。
在神经退行性疾病中,病理过程的核心特征往往是蛋白质的错误折叠和聚集。研究表明,如TDP-43和FUS等与ALS相关的蛋白,其正常的生理功能依赖于形成动态的液态凝聚体。但在疾病状态下,这些凝聚体变得异常粘稠,最终“老化”成固态的、有毒性的包涵体,破坏神经元功能【6】。靶向并恢复这些凝聚体的正常流动性,成为治疗此类疾病的全新策略。
3. 凝聚体功能失调:多种重大疾病的共同根源
针对生物分子凝聚体开发的药物被称为“凝聚体调节剂”(condensate-modulating drugs, c-mods)。2022 年,Mitrea等人在Nature Reviews Drug Discovery上正式提出这一概念,将其定义为通过调节特定生物分子凝聚体的物理性质、大分子网络、组成、动态学和/或功能来预防或逆转疾病的药物【2】。
3.1 核心逻辑与传统药物的区别
c-mods的核心逻辑与传统药物存在本质区别。传统小分子药物依赖于与靶蛋白折叠结构域中特定结合口袋的高亲和力结合(通常需要nM级别的Kd);而c-mods的作用对象是凝聚体这一涌现性(emergent)结构——凝聚体的性质并非由单一蛋白决定,而是由数百种生物分子通过弱多价相互作用共同构成的网络所决定。因此,c-mods不需要完全抑制某一蛋白的活性,而是通过对弱相互作用网络的微扰来改变凝聚体的整体行为,这为靶向IDR(内在无序区)等传统难成药靶点提供了全新可能。
3.2 四类作用机制分类
根据Patel等人(2022)提出的基于可观测表型的分类框架,c-mods可分为四大类【12】:
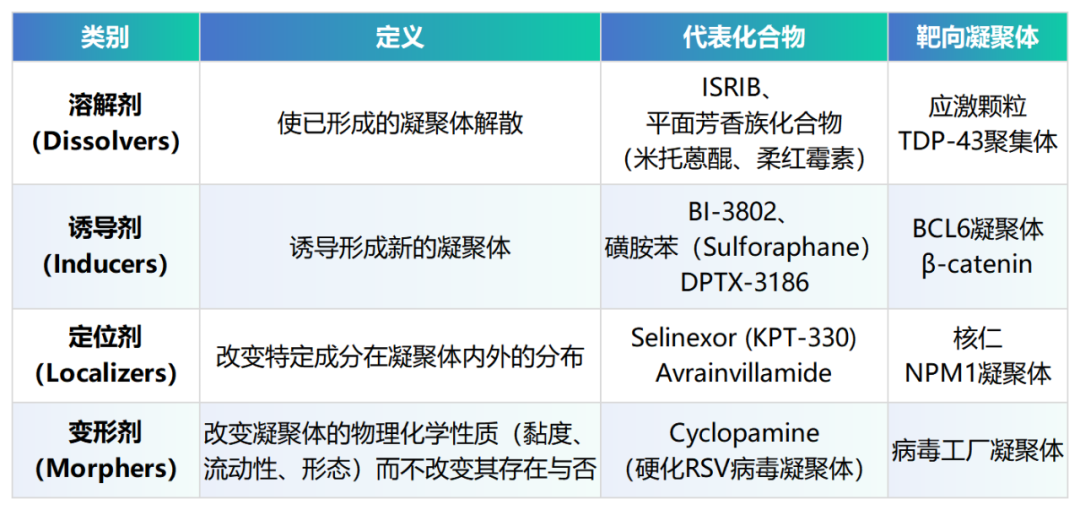
这四类效果并非互斥,同一化合物可能同时具有多种效果,且任一效果都可能对疾病产生治疗意义。
4. c-mods筛选的核心技术体系
c-mods的发现高度依赖于能够量化凝聚体表型的技术平台,这与传统的生化活性筛选(如酶活抑制)存在根本性差异。
4.1 高内涵成像(HCI)表型筛选
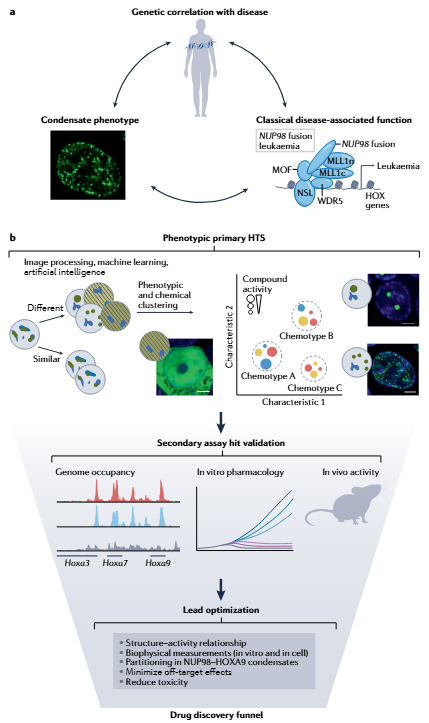
高内涵成像(High-Content Imaging, HCI)是目前c-mods发现的主流筛选方法。其核心原理是:将目标蛋白以荧光标签(如GFP)融合表达于细胞中,利用自动化荧光显微镜对大量细胞进行高通量成像,随后通过图像分析算法对凝聚体的数量、大小、形态、荧光强度、共定位系数等多维参数进行定量分析。Dewpoint Therapeutics的ERSA平台是该方法的最高水平代表:其化合物库超过40万种,已积累近3 PB的凝聚体成像数据(逾400万张细胞图像),并配合专有AI引擎ERSAi™对图像数据进行深度学习分析,识别人眼无法察觉的“隐晦表型”,并为每个化合物生成独特的“数字签名”以解析其作用机制【13】。
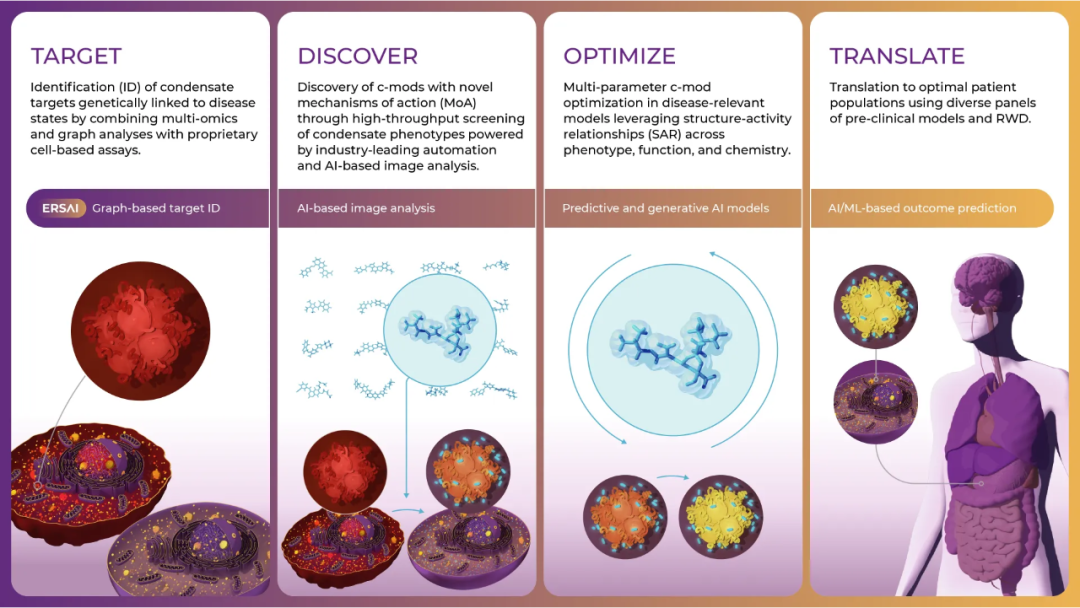
4.2 Corelet™ 光遗传学精准筛选平台
Corelet™技术由Nereid Therapeutics(前身为 Hyman 实验室衍生公司)开发,是目前最精准的凝聚体靶向筛选工具之一【14】。其核心原理是光遗传学控制的相分离:将目标蛋白与光敏蛋白(如iLID)融合,再与铁蛋白纳米笼(ferritin nanocage)偶联,形成“Corelet”。在蓝光照射下,iLID 发生构象变化,驱动目标蛋白在细胞核内迅速聚集,形成人工凝聚体;撤去光照后,凝聚体解散。这种可精准时空控制的手段使研究者能够:
(1) 在生理相关条件下(内源性表达水平)诱导凝聚体形成,避免过表达伪影;
(2) 以凝聚体的形成/解散动力学作为高度灵敏的筛选读出;
(3) 对特定蛋白(如FUS、TDP-43、BRD4等)的凝聚体进行靶向筛选,而非依赖通用表型。截至2025年,Nereid已利用Corelet平台筛选了数十万种小分子,获得了多个具有凝聚体调节活性的先导化合物【14】。
4.3 其他关键技术
- 体外重构凝聚体筛选:在试管中诱导纯化蛋白发生相分离,直接测量化合物对热力学平衡的影响,排除细胞内复杂背景干扰。在α-synuclein的c-mod发现中,Dada等人(2024)正是通过体外重构体系,系统筛选了多种氨基固醇类化合物,发现claramine能够稳定α-synuclein液滴并抑制其向淀粉样纤维的转变,为帕金森病提供了全新的治疗思路【15】。
- FRAP(荧光漂白恢复)动力学分析:评估凝聚体物质状态(液态vs. 凝胶态/固态)的标准方法,用于验证化合物是否改变了凝聚体的流动性。
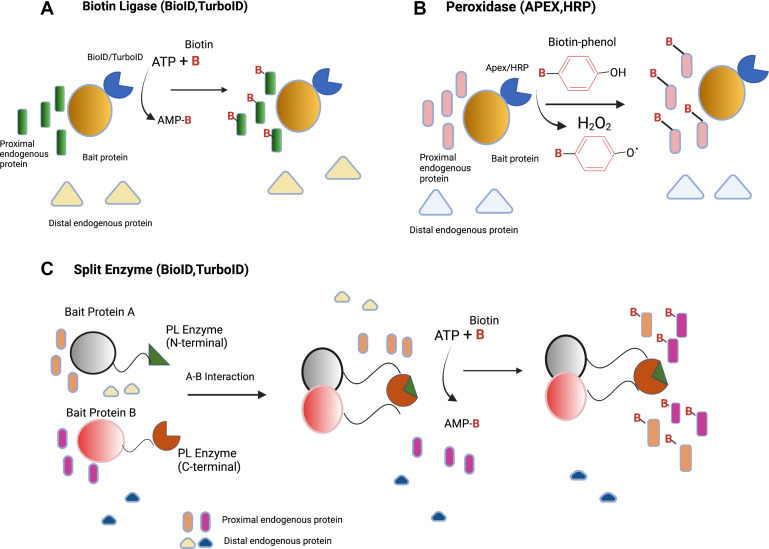
- 邻近标记蛋白质组学:利用BioID、TurboID、APEX2等技术揭示凝聚体的“分子社区”,识别疾病相关的异常组成变化。在NUP98-HOXA9白血病凝聚体的研究中,邻近标记蛋白质组学揭示了其与染色质重塑因子的广泛相互作用网络,为c-mod的靶向设计提供了关键信息【1】。
5. c-mods与凝聚体的分子相互作用原理
理解c-mods如何与凝聚体相互作用,是进行理性设计和化学优化的基础。
5.1 凝聚体富集效应(Condensate Partitioning)
凝聚体具有独特的理化环境。Nature Chemistry(2024)的一项系统性研究对约 1700种生物活性小分子在不同凝聚体中的分配系数进行了定量测定【18】,发现:
- 芳香性(aromaticity)和平面性(planarity)是预测高分配系数的最重要特征,这与凝聚体内IDR中酪氨酸(Tyr)、苯丙氨酸(Phe)等芳香族残基的高密度密切相关;
- π-π堆叠和阳离子-π相互作用是小分子在凝聚体内富集的主要驱动力;
- 亲水性过强(高LogP)或分子量过大的化合物往往分配系数较低;
- 不同凝聚体(如应激颗粒vs. 转录凝聚体)对小分子的选择性不同,这为开发凝聚体选择性c-mods提供了可能【2】。
通过优化这些性质,可以主动提升药物在特定凝聚体中的富集程度,从而在更低的系统给药浓度下实现有效的凝聚体内药物浓度,降低脱靶毒性。
5.2 IDR 靶向策略
针对缺乏稳定结构的IDR,目前的c-mod设计策略包括:
- 共价结合策略:利用IDR中半胱氨酸(Cys)等亲核残基,设计共价弹头(covalent warhead),实现对IDR的不可逆结合。BAY 249716和BAY 1892005是靶向p53 IDR的共价结合剂,可诱导或溶解p53突变体凝聚体。MYC IDR的共价小分子结合剂也已被报道【16】。
- 稳定无活性构象策略:通过弱相互作用稳定IDR的无活性构象,降低其参与相分离的能力。EPI-001系列化合物通过与雄激素受体IDR结合,溶解雄激素受体转录凝聚体,已进入临床试验【2】。
- 多价相互作用竞争策略:IDR通过多价弱相互作用驱动相分离,低价竞争性配体可以通过“封端”IDR的相互作用位点,降低其有效价态(valency),从而抑制凝聚体形成。短诱饵RNA(bait RNA)通过竞争TDP-43的RNA结合位点,抑制了TDP-43包涵体的形成【2】。

6. c-mods 通用研发路径:五阶段框架
基于上述技术体系和机制原理,结合Mitrea 等人(2022)提出的c-mod发现管线框架以及Dewpoint、Nereid等公司的实践经验【2】,可以归纳出一套适用于任意相分离蛋白靶点的通用五阶段研发路径:
第一阶段:凝聚体假说建立与靶点验证
核心问题:目标蛋白是否在疾病相关细胞中形成异常凝聚体?该凝聚体是否与疾病表型存在因果关系?
关键步骤:
- 生物信息学预测:利用CatGranule、P-score、PLAAC等算法预测目标蛋白的相分离倾向;结合D2P2、MobiDB等数据库分析IDR区域;利用PhaSepDB、DrLLPS等相分离蛋白数据库检索已知信息。AlphaFold预测结果中的“无序/受挫折叠”区域可提供重要线索。
- 细胞内凝聚体可视化:通过内源性荧光标记(CRISPR敲入GFP标签)或过表达荧光融合蛋白,在疾病相关细胞系(患者来源iPSC、原代细胞)中观察目标蛋白的亚细胞定位和凝聚体形成情况。
- 凝聚体-疾病表型关联验证:通过遗传学手段(IDR截短突变、相分离缺陷突变)证明凝聚体形成与疾病表型(如基因转录异常、蛋白功能丧失、细胞死亡)之间的因果关系。
- 凝聚体组成表征:利用邻近标记蛋白质组学(TurboID/APEX2)绘制凝聚体的蛋白质组图谱,识别关键支架蛋白(scaffold)和客户蛋白(client),为后续筛选和机制研究提供靶标清单。
典型案例——NUP98-HOXA9(AML):人类遗传学数据显示NUP98融合癌基因与AML强相关;细胞实验证明NUP98-HOXA9通过FG重复IDR在细胞核内形成异常凝聚体;FRAP显示凝聚体内分子在秒级时间尺度内快速交换;ChIP-seq证明凝聚体与HOX基因簇的异常转录直接相关【17】。
第二阶段:凝聚体表征与筛选模型建立
核心问题:如何建立可靠的、与疾病功能读出相关联的凝聚体表型检测体系?
关键步骤:
- 体外重构体系建立:纯化目标蛋白(包含IDR),在生理条件下诱导LLPS,建立体外液滴模型。系统测定相分离的热力学参数(临界浓度Csat、相图)和动力学参数(液滴融合速率、FRAP恢复时间)。
- 细胞内凝聚体定量分析方法建立:开发基于HCI的多参数凝聚体定量分析方法,确定关键表型参数(如凝聚体数量/大小/荧光强度/形态)及其与功能读出(如靶基因转录水平、细胞存活率)的相关性。
- 筛选模型验证:利用已知的遗传学工具(IDR突变体、相分离缺陷突变体)验证筛选模型的特异性;确定Z'因子(Z' factor)等统计指标,确保筛选模型适合HTS。
- Corelet 模型建立(可选):对于需要精确控制凝聚体形成时机的靶点,可构建Corelet细胞系,实现光控相分离,提高筛选的信噪比。
第三阶段:高通量筛选与苗头化合物发现(HTS & Hit Identification)
核心问题:从化合物库中找到能够改变目标凝聚体表型的苗头化合物(hits)。
筛选策略选择:

苗头化合物过滤与优先化:初步筛选获得的hits 需经过多轮过滤:排除泛筛选干扰化合物(PAINS)、荧光干扰化合物、细胞毒性化合物;通过剂量-效应曲线确认活性;通过正交实验(如体外重构、FRAP)验证凝聚体调节活性;利用AI/ML对hits进行聚类分析,识别不同化学类型(chemotype)和可能的作用机制。
第四阶段:苗头化合物到先导化合物优化(Hit-to-Lead Optimization)
核心问题:如何在保持凝聚体调节活性的同时,优化化合物的效力、选择性和成药性?
多维优化目标:c-mods的先导化合物优化需要同时考量以下维度,这与传统药物优化存在显著差异:
- 凝聚体内富集优化(Partitioning Optimization): 通过调整化合物的芳香性、平面性、氢键供体/受体数量和电荷分布,优化其在目标凝聚体中的分配系数,提高凝聚体内的有效浓度。Nature Chemistry(2024)的系统性研究为此提供了定量指导【18】。
- 凝聚体选择性优化(Condensate Selectivity):不同凝聚体具有不同的微环境(如电荷密度、疏水性、RNA含量),通过调整化合物的理化性质,可以实现对特定凝聚体的选择性富集,降低脱靶效应。
- 功能性读出驱动的SAR:构效关系(SAR)分析不仅依赖于凝聚体表型(如液滴大小/数量),更需要与疾病相关的功能读出(如靶基因转录水平、细胞存活率)相关联,确保化合物优化方向与治疗目标一致。
- 传统ADMET优化:口服生物利用度、代谢稳定性、血脑屏障穿透性(对神经退行性疾病靶点尤为关键)、毒性等传统成药性参数同样需要在此阶段进行优化。
计算辅助优化工具:
- 全原子分子动力学模拟(all-atom MD): 阐明小分子与IDR 的结合模式和构效关系(如 EPI-002 与雄激素受体IDR的结合机制);
- 粗粒化模拟(coarse-grained MD): 模拟小分子对相分离热力学的影响;
- 机器学习预测模型:基于已有数据预测新化合物的凝聚体分配系数和活性【19】。
第五阶段:临床前验证与IND 申报(Preclinical Validation & IND)
核心问题: 候选化合物是否在疾病相关动物模型中展现出有效性和安全性?
关键验证实验:
- 机制验证:在疾病相关细胞系和患者来源细胞(PDC)中验证候选化合物对目标凝聚体的调节作用(凝聚体表型、FRAP 动力学)和功能影响(靶基因表达、蛋白功能)。
- 体内有效性验证:在疾病相关动物模型(CDX/PDX肿瘤模型、神经退行性疾病转基因小鼠模型)中验证口服给药的有效性,监测肿瘤消退、生物标志物变化(如NfL水平)等终点。
- 安全性评估:系统评估候选化合物对正常细胞(尤其是与目标凝聚体相似的正常凝聚体)的影响,建立治疗窗口(therapeutic window)。
- 生物标志物开发:开发可用于临床的药效学生物标志物(PD biomarker),如DPTX-3186使用AXIN2转录水平作为Wnt通路抑制的生物标志物,TDP-43项目使用NfL(神经丝轻链)作为神经损伤的生物标志物。
7. 临床进展与典型案例分析
靶向凝聚体的药物开发已从理论概念迅速走向临床验证。
7.1 Dewpoint Therapeutics 的管线进展
Dewpoint公司利用其AI驱动的平台,建立了一条涵盖肿瘤、神经科学和心血管代谢疾病的丰富产品管线。
7.2 其他典型案例
- α-synuclein → Claramine:通过体外重构筛选发现,Claramine能够稳定α-synuclein液滴(变形剂效果),抑制其向病理性淀粉样纤维的转变【15】。
- NUP98-HOXA9:针对AML中的融合癌蛋白,通过HCI筛选识别出能够溶解异常转录凝聚体的化合物,从而关闭致癌基因的转录【20, 21】。
挑战与未来方向
尽管前景广阔,但凝聚体药物开发仍面临诸多挑战:
- 选择性与安全性:如何特异性靶向病理相关凝聚体而不影响正常生理功能。
- 靶点去卷积:表型筛选获得的hits 往往难以确定其直接分子靶点。
- 临床生物标志物:缺乏直接监测临床患者体内凝聚体状态的手段。
AI驱动的凝聚体预测与c-mod设计:随着凝聚体-化合物相互作用数据的积累,基于深度学习的凝聚体分配系数预测模型(如JACS Au, 2025)和c-mod活性预测模型将日趋成熟。未来方向将聚焦于AI驱动的从序列到候选药物的端到端AI设计【1, 11】;
PROTAC/分子胶水与凝聚体调节的结合:将蛋白降解技术(PROTAC、分子胶水)与c-mod相结合,通过降解凝聚体支架蛋白来彻底消除病理凝聚体,是一个极具潜力的新方向。
多模态c-mods:同时靶向凝聚体的多个组分(如同时靶向支架蛋白和调控酶),或将c-mod与传统靶向治疗联合使用,有望克服单一机制的局限性。
凝聚体靶向递送:利用凝聚体的富集效应,设计能够在特定凝聚体中高度富集的纳米载体或前药,实现对凝聚体内靶点的精准递送。
结 论
生物分子凝聚体的发现是细胞生物学领域的重大突破,它彻底改变了我们对疾病发生机制的理解。通过将药物开发的焦点从单个分子扩展到功能性的分子社区——凝聚体,可以开启了一个全新的治疗维度。随着像 Dewpoint Therapeutics 这样的公司将候选药物推向临床,一个由凝聚体调节剂引领的药物开发新纪元已经到来,它将为全球无数患者带来革命性的治疗方案。